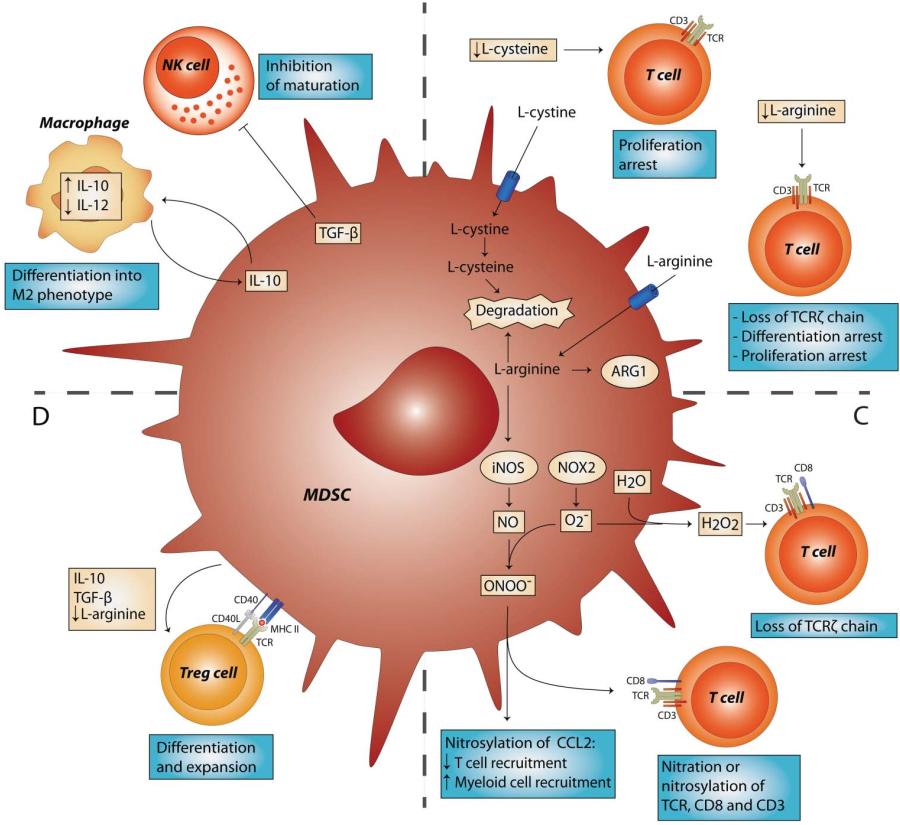
10.4 MDSCs的累积及功能的调控机制
MDSC研究领域的一个主要问题是它们的扩增、积累和激活是如何调节的?
几年前,我们提出了一个双信号模型,该模型描述了MDSC的积累需要两种不同类型的信号。该模型分为两个阶段:(1)扩增阶段,与其终末分化抑制相关;(2)激活阶段,负责将未成熟的髓样细胞转化为免疫抑制的MDSCs。
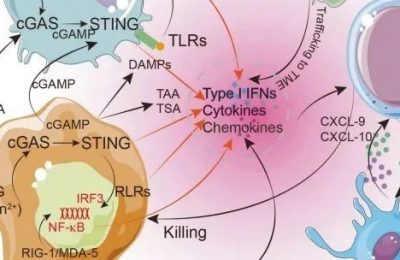
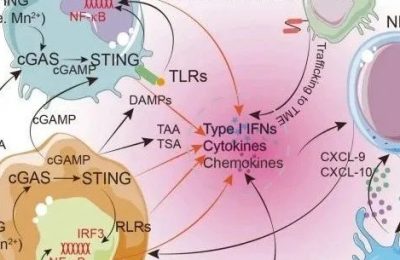
我们认为,这两个阶段部分重叠,但受不同的转录因子和中间产物控制(Fig10.3)。
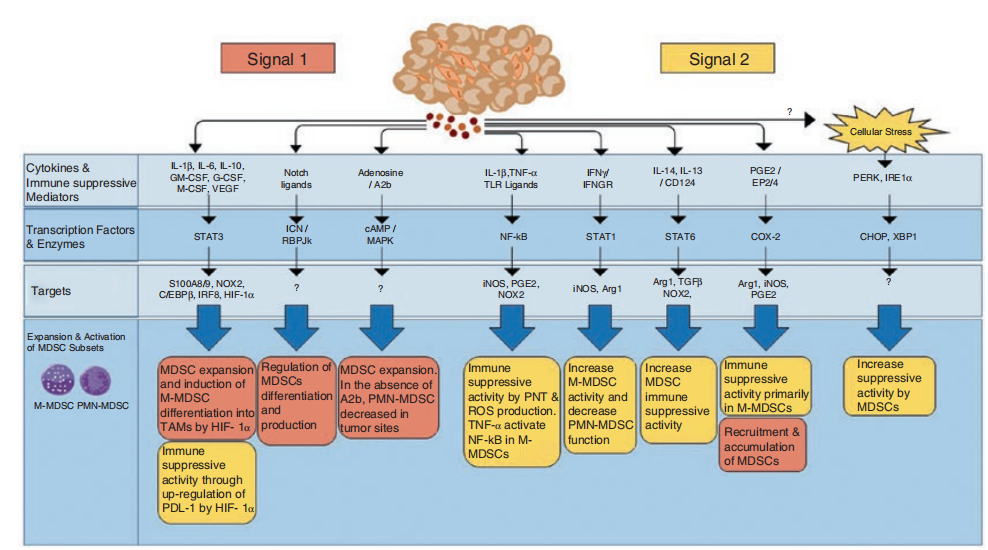
Fig.10.3 肿瘤相关因子和分子通路调节肿瘤中MDSC的扩增和功能。肿瘤微环境产生了大量的细胞因子和免疫抑制介质,信号1和2分别调节MDSC的扩增和免疫抑制活性。MDSC暴露在肿瘤分泌的生长因子(信号1)和其他介质中,调节M-MDSC或PMN-MDSC扩增和聚集过程中涉及的几个信号通路,特别是STAT3。然而,对于未成熟的髓样细胞获得免疫抑制活性,需要信号2来同时产生M-MDSCs和PMN-MDSCs。这些信号通路对于PMN-MDSCs和M-MDSCs(NF-κB、STAT3、STAT6、ER应激反应)都很重要,或者可能在MDSC亚群(STATT1)中具有相反的作用。COX-2对M-MDSCs更具有特异性。
第一阶段主要由肿瘤衍生生长因子以及STAT3、IRF8、C/EBPβ、RB1、NOTCH、腺苷受体A2B和NLRP3驱动。
STAT3
MDSCs中信号转导和转录激活因子3(STAT3)的激活需要髓系特异性生长因子,如GM-CSF、G-CSF、M-CSD、IL-6、VEGF和其他几种因子。
STAT3在MDSCs的调控中起重要作用。例如,用不同的STAT3抑制剂治疗的荷瘤小鼠表现为MDSC积聚减少。
有趣的是,STAT3不仅参与MDSCs的扩增,还参与其功能和分化;例如,M-MDSCs能够通过STAT3的调节在肿瘤微环境中分化为肿瘤相关巨噬细胞(TAMs)。
IRF8
干扰素调节因子8(IRF8)被认为是MDSC分化的负调控因子。没有IRF8的情况下,MDSC在脾脏和肿瘤组织中的积聚增加;然而,在自发的小鼠肿瘤模型中,IRF8的过表达会导致MDSCs积聚减少。
C/EBPβ
CAAT增强子结合蛋白β(C/eBPβ)是其家族中唯一与MDSC扩增相关的成员。缺乏造血细胞特异性C/EBPβ的小鼠的MDSCs数量较少,尤其是M-MDSCs,表明其在MDSCs分化中起作用。
Rb1
视网膜母细胞瘤蛋白1(Rb1)与在小鼠和人类中发现的MDSCs有关。在荷瘤小鼠中,Rb1被证明可以调节M-MDSCs向PMN-MDSCs分化。高Rb1水平的M-MDSCs主要产生巨噬细胞和DC,而Rb1水平低的M-MDSCs绝大多数分化为PMN-MDSCs。最近,在小鼠乳腺癌转基因模型中证实了存在Rb1lo Ly6G+PMN-MDSC的蓄积。
Notch 信号通路
另一条参与MDSCs聚集和分化的途径是Notch信号通路。已有研究表明,在MDSCs中,酪蛋白激酶2(CKII)的激活下调了Notch的表达。
因此,用CKII抑制剂治疗的荷瘤小鼠在Notch信号和DC分化方面表现出改善。这些结果表明,Notch信号的下调使造血干细胞向MDSCs分化而不是向DC分化。
最近,研究表明,Notch 信号的抑制增强了 PMN-MDSCs 的产生,但减少了 M-MDSCs 的产生。
Adenosine Receptor A2B
在肿瘤微环境中,通过与MDSC上表达的腺苷受体A2B结合,细胞外腺苷水平升高,这与MDSC聚集有关。
与野生型相比,缺乏A2B的荷瘤小鼠的MDSCs数量较少,这表明A2B受体在MDSCs,特别是PMN-MDSCs的扩增中起关键作用。
这与最近一项使用A2B激动剂和拮抗剂治疗黑色素瘤模型的研究有关,在该模型中,激动剂治疗增加了肿瘤生长和MDSC积聚,而拮抗剂治疗导致肿瘤进展减缓,肿瘤组织中MDSC数量减少。
NLRP3
NLR3(NLRP3)是一种与炎症小体相关的细胞内感受器,激活后可诱导白细胞介素-1(IL-1)β和IL-18的产生。
肿瘤相关的MDSCs表达NLRP3,与野生型小鼠相比,Nlrp3−/−小鼠肿瘤组织中MDSCs的水平较低,这表明NLRP3可能在MDSCs的扩增和积累中发挥作用。
然而,需要更多的研究来确定NLRP3在MDSC功能中的确切作用。显然,这些信号通路调节MDSC的扩增,并参与阻止正常的未成熟髓系细胞分化。
然而,这些因子中的大多数都与MDSC调节的第一阶段有关,但这也并不足以促进它们被激活成为免疫抑制细胞(Fig10.3)。
作为我们双信号模型的一部分,在MDSCs中发现的抑制活性的产生主要是由肿瘤基质产生的因子介导的,包括促炎分子,如干扰素γ,IL-1β,IL-13,Toll样受体配体等。
这些分子中的每一个都参与了多种信号通路的激活,其中包括NF-κB、STAT1、STAT6、前列腺素E2(PGE2)和环氧合酶(COX-2)、高迁移率族蛋白1(HMGB1)和缺氧诱导因子-1α(HIF-1α),他们都与MDSCs的抑制活性有关。
最近,内质网(ER)应激途径被认为与肿瘤中发现的MDSCs的抑制活性有关。
NF-κB
在MDSCs中,NF-κB通路的激活主要是由TLR配体:IL-1β或TNF-α介导的,IL-1β或TNF-α均被证明能增强抑制活性。
TLR配体在MDSC功能中的作用尚不清楚。一些研究表明,TLR配体驱动MDSCs的抑制活性,而另一些研究表明这些配体抑制其功能。
在炎症和癌症过程中,肿瘤细胞产生的IL-1β通过NF-κB途径激活MDSCs,导致PNT的产生增加。另一个众所周知的NF-κB途径的激活剂是肿瘤坏死因子-α(TNF-α),它与MDSCs的成熟和功能有关。
结果表明,跨膜型TNF-α可通过其受体增强MDSCs的抑制活性,并以NF-κB依赖的方式调节iNOS的表达。
一项使用炎症小鼠模型的研究表明,TNF-α的作用仅限于M-MDSCs。此外, NF-κB通过TNF-α受体激活可以促进MDSCs的存活。目前,NF-κB可以参与骨髓间充质干细胞的扩增,但它的主要作用是发出信号激活这些细胞,从而获得抑制表型,特别是在M-MDSCs中。
STAT1
一些研究表明,STAT1的激活与MDSCs的抑制活性有关。STAT1在干扰素γ刺激下被激活,参与诱导型一氧化氮合酶和精氨酸的表达上调。Sinha等人证明通过干扰素γ激活STAT1对MDSC的积累或功能无调节作用。随后的研究集中在干扰素γ-STAT1通路在MDSC亚群抑制活性中的作用。
最近的一项研究表明,STAT1通过增强M-MDSCs的抑制功能发挥主要作用。然而,通过干扰素γ受体激活STAT1可降低PMN-MDSC的功能和存活率。
这些研究表明,干扰素γ-STAT1信号通路在M-MDSCs和PMN-MDSCs中可能具有相反的作用。
事实上,这可以解释为什么最初的研究没有观察到干扰素γ和/或STAT1参与MDSCs的抑制活性,因为SinHA等人的研究分析了总MDSCs(CD11b+Gr-1+)的功能,而不是单独分析亚群。
在缺少STAT1信号通路的情况下,可能是PMN-MDSC数量较高,而不是M-MDSC数量较高,从而观察到了抑制活性。
STAT6
另一个参与MDSCs功能的转录因子是STAT6。在MDSCs中,STAT6信号通路在IL-4和IL-13的刺激下通过其受体(CD124、IL-4Rα)被激活,导致Arg1表达上调和产生转化生长因子-β。此外,STAT6已被证明参与了MDSCs的存活和积聚。
PGE2
前列腺素,尤其是前列腺素E2,在肿瘤MDSCs的抑制活性产生中起着重要作用。
Rodriguez及其同事发现,通过PGE2受体E-前列腺素(EP)4(在MDSCs中表达)发出的信号可诱导Arg1的表达及其活性。此外,PGE2产生的主要调节因子COX-2的表达与肿瘤浸润性MDSCs中Arg1和iNOS的诱导直接相关。
这些结果得到了OberMajer等人的证实。他们描述了PGE2和COX-2之间的正反馈循环,导致单核细胞转化为M-MDSCs。
在此基础上,利用PGE2诱导抑制因子的产生,产生了体外MDSCs。在小鼠间皮瘤和胶质瘤模型中,用COX-2抑制剂治疗可抑制MDSCs的聚集和功能。
此外,在接受COX-2抑制剂治疗的黑色素瘤患者中,证实了PGE2参与了MDSC的功能,这些患者的MDSC样细胞抑制活性较低。
PGE2在调节MDSCs的免疫抑制方面发挥了作用,但也与MDSCs的募集和积聚有关。
HGMB1
HGMB1是一种DNA结合蛋白,在肿瘤微环境中含量很高。
HGMB1通过与TLR4和晚期糖基化终产物受体(RAGE)结合影响MDSC。通过这两种受体发出的信号激活了NF-κB通路。
Parker等人的研究成果表明,HGMB1通过诱导肿瘤微环境中细胞的自噬来驱动MDSCs的聚集和抑制活性,并促进MDSCs的存活。
最近,Su等人证明,在体内,用抗HGMB1B盒的单克隆抗体阻断HGMB1可以减少M-MDSCs在脾脏和肿瘤组织中的积聚。这些研究表明,HGMB1可能在MDSCs的积累和功能中起关键作用。
HIF1α
无氧(缺氧)是肿瘤微环境与外周淋巴器官的最大区别之一。
研究表明,在肿瘤微环境中,缺氧诱导因子-1α能够以STAT3依赖的方式调节MDSC的功能和M-MDSCs向TAMs的分化。
最近,诺曼等人提出缺氧诱导因子-1α选择性地上调程序性死亡配体1(PD-L1),这是MDSC上的一种免疫检查点配体 (Fig10.3)。
最近的研究表明,内质网应激反应参与了MDSCs在荷瘤宿主中的抑制行为。
内质网应激反应途径,也被称为未折叠蛋白反应(UPR),负责外在或内在应激过程中维持内质网稳态。
内质网应激反应由三个内质网定位的跨膜蛋白传感器组成:肌醇需要酶1α(IRE1α)、激活转录因子6α(ATF6α)和双链RNA依赖激酶(PKR)样ER相关激酶(PERK)。
这些ER感受器的激活导致一些转录因子的上调,包括剪接的X-box蛋白1(sXBP1)、CCAA T增强子结合蛋白同源蛋白(CHOP)等。
从荷瘤小鼠和癌症患者身上分离的MDSCs都过表达几个ER应激的标志物,包sXBP1和CHOP,并伴随ER增大(ER应激的一个标志)。
用内质网应激诱导剂thapsigargin治疗的荷瘤小鼠,其MDSCs的数量增加,具有强大的抑制活性。我们发现,用thapsigargin诱导内质网应激以IRE1α/XBP1依赖的方式将正常人中性粒细胞转化为抑制的PMN-MDSC表型。
除了IRE1α/XBP1在MDSC功能中的作用外,最近的研究发现CHOP还参与了肿瘤相关MDSCs的抑制活性。
例如,CHOP缺陷的MDSCs失去了抑制功能,获得了免疫刺激表型。尽管CHOP缺陷的MDSCs不能抑制以非抗原特异性方式刺激的T细胞,但它们保留了阻断抗原特异性T细胞的能力。
目前,只有IRE1α/XBP1和PERK/CHOP信号通路参与了MDSCs的功能。然而,需要更多的研究来了解内质网应激反应调节MDSCs抑制活性的具体机制。
10.5 MDSCs与其他髓系细胞的关系
多年来,我们对癌症中髓系细胞的了解揭示了它们在肿瘤发生、发展和转移中的重要作用。终末分化的髓系细胞主要有巨噬细胞、树突状细胞和中性粒细胞。
现已清楚的是,肿瘤微环境通过将髓系细胞阻滞在具有强大免疫抑制活性的未成熟阶段来改变髓系细胞的分化。
另一个参与免疫抑制的髓系细胞是肿瘤相关巨噬细胞(TAM),它来源于肿瘤微环境中的肿瘤浸润性单核细胞或M-MDSC。然而,MDSCs与其他髓系细胞的关系尚不清楚。
现面临的主要问题是缺乏MDSCs的特异性标记物,这使得很难准确地将这些细胞与单核细胞和中性粒细胞区分开。这就是为什么在之前的许多研究中,具有抑制活性的MDSC样细胞被称为单核细胞和中性粒细胞。
最近的数据已经为MDSCs在癌症中的特殊性质提供了证据,免疫抑制活性是MDSCs的固有特性。例如,在肿瘤分泌因子存在的情况下,骨髓祖细胞可以在体外产生MDSCs。
与MDSCs不同,成熟的中性粒细胞或单核细胞在存在这些肿瘤相关因子的情况下,在体外不能抑制T细胞反应。
几项涉及基因组、蛋白质组和转录组分析的研究已经提供了证据,支持荷瘤小鼠PMN-MDSCs和中性粒细胞之间的差异。
此外,最近的一项研究提供的信息表明,根据PMN-MDSCs的基因图谱,PMN-MDSCs不同于健康捐赠者或癌症患者的中性粒细胞。在人类中,PMN-MDSCs可以通过LOX-1的表达与中性粒细胞区分开来;在小鼠中,LOX-1的作用尚不清楚。
在缺氧的肿瘤微环境中,M-MDSCs分化为TAMs,这受HIF-1α和STAT3激活的调节。两种髓样细胞都有很强的免疫抑制功能,特别是在肿瘤部位;然而,M-MDSCs和TAMs可以通过单核/巨噬细胞相关标志物的变化来区分。
在M-MDSC分化为TAMs的过程中,主要观察到F4/F80和CD115表达增加,Ly6C表达减弱,IRF8和S100A9蛋白表达降低。
MDSCs具有中性粒细胞和单核细胞所没有的一些生化特征,包括高Arg1和iNOS的表达和活性,以及高水平的ROS和PNT产生。
此外,最近的研究表明,MDSCs的内质网应激反应增加,这与其抑制功能有关。
在癌症中不同类型的髓系细胞当中,目前对M-MDSCs和TAMs之间的关系了解的更多一些。最近几个小组的数据显示,M-MDSCs是肿瘤微环境中的TAM前体细胞。
此外,巨噬细胞有两种极化表型:具有抗肿瘤特性的M1型巨噬细胞和具有促肿瘤特性的M2巨噬细胞。后者与TAMs关系更密切,但尚不清楚这两种表型是否是同一种巨噬细胞的表型,以及在肿瘤微环境中调节它们的分子机制是什么。
此外,在TAM分化之前,巨噬细胞的极化是否由M-MDSCs决定尚不清楚。另一方面,PMN-MDSCs的性质及其与中性粒细胞的关系也是不同研究持续争论的主题。
虽然PMN-MDSCs在该领域被广泛接受,但癌症中性粒细胞极化的概念提出了这样的问题:这些细胞在病理上是否与PMN-MDSCs相似?
与巨噬细胞相似,肿瘤相关中性粒细胞(TANs)的概念由肿瘤微环境中具有抗肿瘤(N1细胞)或亲肿瘤(N2细胞)特性的中性粒细胞组成。研究表明转化生长因子-β和1型干扰素(IFN)是调节癌症TAN可塑性的细胞因子。
根据定义,中性粒细胞是短暂的终末分化细胞,而PMN-MDSCs是未成熟的细胞。因此,很难想象TANS会在肿瘤微环境中被极化。
事实上,几项研究表明,这些细胞有能力抑制荷瘤小鼠的T细胞反应,这支持了这些细胞是PMN-MDSCs的观点。
然而,TANs的抗肿瘤作用已经在癌症患者中有过描述。最近,Eruslanov等人观察到早期肺癌患者的TANs不是免疫抑制,而是刺激T细胞反应,这项研究为TANS在肿瘤发生的早期阶段的作用提供了证据。
很少有研究涉及MDSCs在肿瘤发生中的作用,以及它在早期阶段如何调节免疫反应。
例如,Ortiz等人研究表明,小鼠暴露在香烟烟雾中会导致不同器官中非抑制性MDSC样细胞的积累。然而,当香烟烟雾和致癌物联合起来促进肺癌的发展时,MDSCs表现出很强的免疫抑制活性,这些数据表明MDSC的功能受肿瘤微环境的控制。
我们对MDSCs的性质以及它们在肿瘤发展过程中与其他髓系细胞的关系的了解将为肿瘤提供新的治疗思路。
10.6 靶向MDSCs的肿瘤治疗策略
有充分的证据表明,MDSC在外周淋巴器官和肿瘤组织中的积聚与癌症患者不良的预后和临床结果相关。
此外,MDSCs与抗癌治疗有关,包括受体酪氨酸激酶抑制剂Sunitinib以及几种肺癌和多发性骨髓瘤的化疗药物。
因为这些细胞具有抑制抗肿瘤免疫反应的能力,所以包括免疫治疗在内的癌症治疗的效果将取决于MDSCs的免疫抑制作用。
事实上,一些临床研究表明MDSC水平与对免疫检查点阻断剂ipilimumab(抗CTLA-4)和nivolumab(抗PD-1)的反应相关。
目前,随着对肿瘤中MDSCs聚集和功能的分子机制的了解逐渐增加,使得针对这一细胞群体的治疗的可能性大大提高。
针对MDSCs的一些策略包括(A)消除MDSCs,(B)阻止MDSCs的积累,以及(C)灭活MDSCs (Fig10.4)。其中几种策略已经开发出来,目前正在临床上进行测试。
消除MDSCs可以增强包括过继性T细胞转移在内的癌症免疫治疗的抗肿瘤效果。
传统的抗癌药除了对癌细胞有直接作用外,还能使肿瘤宿主中MDSCs耗竭。吉西他滨是第一种被报道能够在肿瘤模型中消除MDSCs的化疗药物。这项研究表明,吉西他滨清除MDSCs可以改善抗肿瘤反应,并增强免疫治疗的效果,从而导致肿瘤消退。
其他显示消除MDSCs的化疗药物包括5-氟尿嘧啶、顺铂、阿霉素等。
另一种消除MDSCs的策略是以肿瘤坏死因子相关的凋亡诱导配体受体(TRAIL-R)为靶点。结果表明,TRAIL-R,尤其是DR5的上调调节了肿瘤宿主中MDSC的存活,并且通过使用TRAIL-R激动剂,MDSC被选择性地消除,导致肿瘤生长以CD8+T细胞依赖的方式减少。一期试验的结果显示,获得的数据支持在癌症患者中使用TRAIL-R激动剂。
最近,一种新的靶向MDSCs的治疗方法被开发出来,它是通过将S100A9衍生的多肽与抗体Fc部分进行基因融合来产生多肽抗体。这些多肽抗体成功地耗尽了小鼠模型血液、脾脏和肿瘤中的MDSCs。
然而,未来的研究需要进一步阐明导致MDSC消除的作用机制(Fig10.4)。
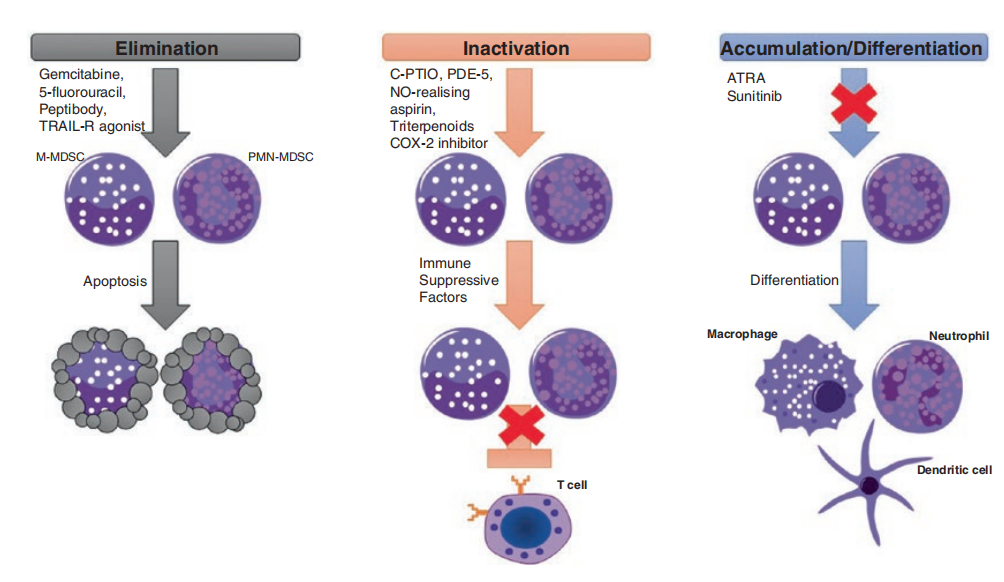
Fig. 10.4 针对MDSCs的治疗策略。靶向MDSCs的策略主要包括消除、灭活或阻断MDSCs的聚集和诱导分化。首先,临床前和临床研究表明,小剂量化疗药物和TRAIL激动剂可以消除MDSC,导致MDSC细胞死亡。其次,几项研究已经证明,通过靶向MDSCs的免疫抑制机制,可以在体内灭活MDSCs。PDE5、NO-realizing阿司匹林、三萜类和COX-2抑制剂通过降低ROS、RNS和精氨酸酶水平来抑制MDSC的功能。最后,可以通过诱导MDSC分化为具有免疫刺激活性的终末分化细胞来抑制MDSC的聚集。
MDSCs利用几种机制来抑制抗肿瘤免疫反应,针对MDSCs的抑制机制已经在癌症患者中进行了测试。
MDSCs产生的ROS和NO的增加在抑制CD8+T细胞应答中起重要作用。活性氧清除剂N-乙酰半胱氨酸(NAC)和过氧化氢酶通常用于体外检测小鼠和人MDSCs的功能。
NRF2是一种转录因子,参与激活抗氧化反应,保护细胞免受ROS的损伤。一种合成的三萜类化合物上调NRF2能够通过减少ROS的产生并抑制其体外抑制功能来中和人类MDSC的活性。
然而,Beury等人最近的一项研究结果显示,在两种小鼠模型中,与NRF2+/+MDSCs相比,NRF2−/−MDSCs的抑制活性降低。
NO清除剂,如羧基PTIO(C-PTIO),最近已经被用于检测MDSCs。在荷瘤小鼠中,C-PTIO治疗降低了MDSCs的功能,提高了过继性T细胞转移的效率。
除阻断ROS和NO外,另一种灭活MDSC功能的方法是抑制MDSC上调的分解代谢酶,包括Arg1和iNOS,它们与抑制T细胞的激活和增殖有关。Arg1和iNOS抑制剂已广泛用于MDSCs的功能检测。
Rodriguez等人的早期研究表明,一种针对Arg1的特异性抑制剂,n -羟基- nor-l-Arg (nor-NOHA),可以抑制肿瘤生长,并降低MDSCs中的Arg1水平。精氨酸(Arg1)抑制剂和诱导型一氧化氮合酶(INOS)抑制剂l-NG-单甲基-l-精氨酸(l-NMMA)在体外均能降低MDSC功能,促进T细胞增殖。
此外,癌症患者PBMCs中的Arg1和iNOS水平已显示出对磷酸二酯酶-5(PDE5)的反应降低。几种PDE5抑制剂已经在临床上用于治疗癌症和其他疾病,可使MDSC数量减少。
最近,一份临床报告表明,接受PDE5抑制剂他达拉非治疗的头颈部癌症患者,MDSCs减少,Arg1和iNOS表达减少,T细胞增加。
一项平行研究表明,他达拉非的治疗不仅减少了MDSCs,还减少了头颈部鳞癌患者血液和肿瘤中的Tregs。此外,这些接受PDE5抑制剂治疗的患者肿瘤特异性CD8+T细胞增加。
硝基阿司匹林,是一种释放NO的阿司匹林,已经被证明在结肠癌模型中诱导MDSCs中的Arg1、iNOS和PNT下调。由于硝基阿司匹林作为过继性T细胞转移的佐剂效果不佳,Molon等人开发了一种T38([3-(氨基羰基)呋喃-4-基]水杨酸甲酯)来抑制MDSCs中的活性氮物质。
CCL2硝化导致肿瘤微环境内CD8+T细胞增加。MDSCs免疫抑制的另一个主要机制是通过激活COX-2产生PGE2。几种COX-2抑制剂,包括塞来昔布,已经在不同小鼠模型的MDSCs中显示出PGE2的产生减少以及Arg1和iNOS的抑制。
塞来昔布选择性抑制COX-2可减少间皮瘤模型中MDSC的数量和ROS和NO的产生,并改进了基于树突状细胞的免疫治疗 (Fig10.4)。
除了清除MDSCs或针对其免疫抑制机制,阻断MDSCs的扩增和诱导分化是另一种有潜力的治疗策略。
众所周知,肿瘤分泌因子在阻止MDSCs分化为树突状细胞或巨噬细胞方面起着关键作用。维生素A的天然氧化代谢物全反式维甲酸(ATRA)在体外和体内诱导MDSCs分化为成熟的髓样细胞的研究也有一些进展。
接受ATRA治疗的癌症患者的髓系/淋巴系树突状细胞比率和免疫反应均有改善。ATRA诱导MDSC分化的机制涉及ERK1/ERK2 MAPK激酶的激活,该激酶上调谷胱甘肽合成酶的表达,增加谷胱甘肽的合成。ATRA在MDSCs中积累谷胱甘肽,降低ROS水平,导致MDSC分化为成熟的髓样细胞。在广泛期小细胞肺癌患者中,ATRA通过耗尽MDSCs增强了癌症疫苗的效果。此外,在肉瘤模型中,ATRA治疗显示MDSC水平和功能降低,改善嵌合抗原受体(CAR)治疗反应。
总结
在过去的10年里,MDSC研究领域获得了更多的关注。MDSCs对于调节癌症和其他病理条件下的免疫反应至关重要。这些细胞可能成为癌症患者选择免疫疗法或抗癌疗法的有力生物标记物。此外,清除癌症患者的MDSCs可以改善最近和即将进行的癌症免疫疗法的效果。
— THE END —
▉ 强烈推荐
▉ OncoLab实验室网站
本公众号上的往期文章同步发布至对应网站OncoLab实验室。
网站自带检索功能,可以根据关键词进行检索,并且可以根据日期及内容分类进行查看 ,大家可以收藏方便在电脑上查看。
网址是:oncolab.cn
▉ OncoLab学术导航
此外,梳理了一下这几年攒的收藏夹,做了一个导航网页,包含常用网站、文献阅读、试剂订购、基金相关、实用工具、常用数据库等分类内容,并且整合了百度、谷歌、必应三大搜索引擎到检索工具中,欢迎收藏或设置为主页使用~
网址是:dh.oncolab.cn
▉ OncoLab知识星球
OncoLab学术星球现已开通,在学习本公众号内容的过程中如果有什么需要讨论交流的地方可以在星球发表留言,也可以分享一下自己的学习心得体会,其他小伙伴看到了可以积极留言回复,我也会积极参与其中,并时常放一些学习资料在上面,希望大家能够在积极交流互帮互助中共同进步~
该星球用于OncoLab公众号读者交流学习使用,永久免费。

关注本号~

加入读者交流群~

加入知识星球~
用于OncoLab读者交流互助永久免费
本篇文章来源于微信公众号: OncoLab
 微信扫一扫打赏
微信扫一扫打赏
 支付宝扫一扫打赏
支付宝扫一扫打赏