32.1 引言
在过去几年中,肿瘤学领域的药物开发专家们经历了一场针对一期临床试验评估药物分子的革命。回顾2010年以前,针对免疫系统的治疗方法只占药物研发的一小部分。然而,免疫检查点抑制剂在多种固体肿瘤治疗中的成功带动了免疫疗法的迅猛发展,无论是作为单药治疗还是联合疗法。这种发展挑战了传统的一期试验设计,催生了创新性及前所未有的药物开发策略。在加速药物开发的过程中,需求空前紧迫,但同时优化早期试验设计并确保患者安全仍是首要任务。本章节将详述免疫治疗时代一期试验的特点,特别是对免疫检查点抑制剂的研究,为早期药物开发提供重要的实践指导。
32.2 针对免疫检查点的抗体治疗
32.2.1 剂量限制毒性与最大耐受剂量(MTD)的确定
在传统的药物开发中,常规的一期临床试验设计遵循“剂量越高越好”的原则,基于假设:更高剂量的抗癌药物能带来更佳疗效,哪怕需要承受较高的毒性(Fig. 32.1)。因此,建立了逐步增加剂量的方案,目的是:(1)限制接受低剂量(可能无效)治疗的患者数量,(2)通过快速递增剂量以缩短试验持续时间,(3)尽可能发现并评估可能对患者造成严重危害的毒性,以此调整药物的风险与收益比。这种不可接受的毒性被称为“剂量限制毒性”(DLT),它指的是在超过预定患者数量的情况下发生的,通常是治疗第一周期(即前3到4周)内出现的3级或以上的严重毒性。根据美国国家癌症研究所不良事件通用术语标准(NCI-CTCAE v4.0),DLT用于界定最大耐受剂量(MTD)。传统的“3+3”剂量递增设计旨在控制DLT发生率在17%-33%之间(即每六名患者中约有一人或更少人出现DLT),从而确定MTD。免疫治疗领域的发展对这种传统药物开发方法提出了新的挑战,尤其是在针对免疫检查点的抗体研究中(详见“Challenges of phase 1 clinical trials evaluating immune checkpoint-targeted antibodies”一文)。
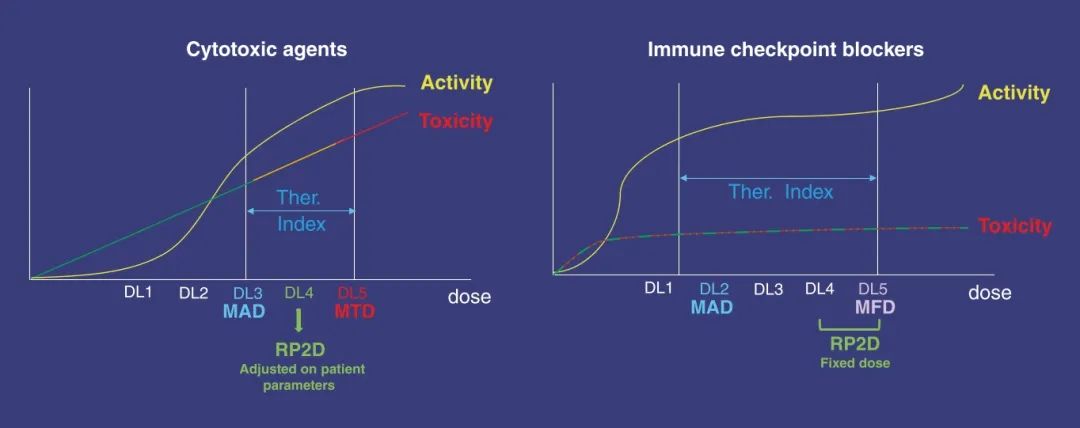
Fig 32.1 免疫检查点阻滞剂的剂量-毒性和剂量-效果关系分析。与传统的细胞毒性药物(如左图所示)不同,免疫检查点阻滞剂(如右图所示)并未展现出任何线性的剂量-毒性或剂量-效果关系。在所有剂量水平上,都可能观察到罕见但严重甚至有时威胁生命的免疫相关不良事件(irAEs)。这些生物活性剂的作用不受超过激发免疫反应所需阈值的剂量影响。因此,这些治疗方法的治疗窗口相比传统细胞毒性药物更为宽广。在图中使用颜色编码表示不同程度的毒性:轻度毒性用绿色线条表示,中度毒性用橙色线条表示,而重度毒性则用红色线条表示。红色点标示出在免疫疗法的剂量依赖性轻度不良事件路径上可能出现的严重免疫相关不良事件(irAEs)。活动程度以黄色表示。图中还标注了几个重要的药物剂量定义:DL为剂量水平、MAD为最小活性剂量、MFD为最大可行剂量、MTD为最大耐受剂量、RP2D为推荐的二期剂量、Ther Index为治疗指数。
32.2.1.1 安全性和最大耐受剂量(MTD)的定义
免疫疗法的毒性表现与传统的细胞毒性疗法或靶向药物相比,在多个方面存在显著差异。例如,在评估抗CTLA-4药物时,已经观察到明确的剂量-毒性关系,然而,在对抗PD-1或抗PD-L1药物进行的一期单药治疗试验中,并未发现剂量与安全性之间有明显的相关性,这一点在后续的研究中也得到了证实。以纳武利尤单抗(Opdivo®, 百时美施贵宝)为例,其疗效和安全性在每2周或每3周给药2至20 mg/kg的剂量范围内表现出相对稳定性。此外,在对13个主要针对抗CTLA-4、抗PD-1或抗PD-L1药物的一期单药治疗试验的考察中,仅有一项试验根据协议定义了剂量限制毒性(DLT),而几乎所有试验都未能确定最大耐受剂量(MTD)。因此,通常建议的第二阶段剂量(RP2D)往往被定为最大可行剂量(MFD),而不是基于安全性数据确定。值得注意的是,纳武利尤单抗的推荐剂量是基于一项大型1b期研究,通过综合药代动力学(PK,反映药物暴露)、药效学(PD)、安全性和有效性数据来确定的。与靶向治疗相似,大多数免疫检查点阻滞剂最终采用固定剂量给药,不根据体重、体表面积或任何患者特定参数进行调整(Fig 32.1)。
在讨论是否存在剂量限制毒性(DLT)或最大耐受剂量(MTD)时,不能简单地认为这些疗法无毒性。免疫检查点阻滞剂引起的严重毒性确实通常低于细胞毒性或靶向疗法,但DLT的缺失主要归因于毒性的不同动力学特征。DLT通常只在治疗的第一个周期评估,这期间观察到的毒性直接关联于药物对健康细胞的急性影响。相较之下,免疫疗法引发的毒性往往是间接的:主要是由于免疫系统对药物的不充分或不适当反应所致,可能是反应的性质或强度不足,或是免疫细胞响应的再编程不当。
这种区别导致了不良事件(AEs)与免疫相关不良事件(irAEs)之间的差异。irAEs通常有一段必要的潜伏期,这个潜伏期随药物的性质而异,如ipilimumab大约为8至10周。因此,仅限于第一个周期的DLT无法充分捕捉到这些与药物相关的潜在毒性,且在剂量增加过程中这些毒性未被充分考虑。考虑到剂量-毒性关系经常缺失和一期试验的迅速完成的必要性,这种晚发性毒性应在一期研究中详细报告并指明其发生周期。例如,在评估抗PD-L1药物如BMS-936559和durvalumab(阿斯利康)的一期试验中,研究者已经选择将DLT评估周期延长至两个周期,以更好地考虑免疫相关的毒性。
在评估免疫疗法的一期试验中,DLT可能不仅局限于严重毒性(NCI-CTCAE 3级以上)。事实上,在更严重的情况下,irAEs需要迅速且特定的治疗,主要依赖免疫抑制和抗炎药物,如类固醇、抗TNF或抗IL-6药物。因此,大多数试验现在建议,一旦观察到中度(2级)irAEs,应立即停止给药,并根据专用指南开始迅速治疗干预。尽管在免疫检查点阻滞剂的药代动力学方面仍存在一些不确定性,但药物中断可能导致药物暴露减少,根据最新的建议,这应被评定为DLT。最重要的是,这种延迟性毒性应在剂量推荐过程以及毒性管理指南的制定中得到特别考虑。
32.2.1.2 剂量时间表
在免疫治疗领域,多数情况下并未观察到明显的线性剂量-反应关系(Fig 32.1)。例如,针对多个剂量水平的nivolumab一期试验中,活性在所有剂量级别上均有观察到;类似地,在转移性黑色素瘤患者中,高剂量的抗CTLA-4药物,如ipilimumab和tremelimumab,并未展示出更高的缓解率。甚至在一项探索三种不同剂量ipilimumab的二期试验中提出的剂量响应,在更大规模的三期试验中也未能得到证实。因此,追求最高可耐受剂量对这些药物来说可能无关紧要,而应考虑它们在较低剂量水平的潜在疗效。在这种背景下,应采用系统的创新设计,允许剂量迅速提高(例如采用加速滴定设计或修改的毒性概率区间设计),并在确定满意的药代动力学和药效学参数后,立即在低剂量水平进行扩展(Fig 32.2)。
除了剂量问题外,最佳给药时间表也存在争议。尽管当前大多数单克隆抗体的给药频率为每2或3周一次,但根据抗体的同型,未能发现一致的模式。还有一些待探索的未知因素,如抗体对其靶标的清除机制。一期临床试验应致力于深入探索这些问题(详见药代动力学和药效学参数部分)。例如,目前抗PD-1和抗PD-L1药物是按连续方案单药治疗实施的,而抗CTLA-4药物ipilimumab已被批准以3 mg/kg剂量,每3周一次,共给予4剂。有研究在评估较低剂量维持治疗的潜力。对于那些在停止治疗后仍显示持续肿瘤反应但最终复发的患者,有效的再诱导治疗潜力也应该被系统考虑。然而,虽然一期试验是测试这些方案的可行性和安全性的平台,真正的疗效比较则需要在后期试验中,针对特定肿瘤类型进行有目的的详细评估。
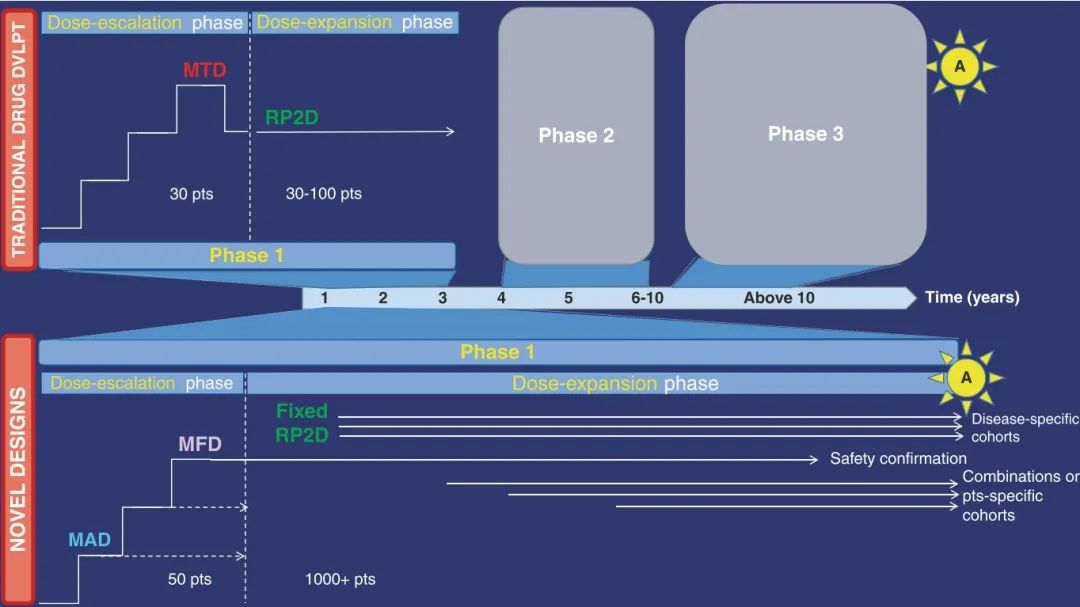
Fig. 32.2 免疫肿瘤学时代的新一期试验设计。传统的药物开发流程(如顶部面板所示)通常包括三个独立的阶段:一期、二期和三期研究(以太阳形状的“A”标识)。传统的一期试验初期为剂量递增阶段,随后是单一队列的剂量扩展阶段,主要目的是评估安全性并确定推荐的二期剂量。在数据支持初步疗效的基础上,约三年的一期试验通常会进入后续的二期和三期试验,整个药物开发周期通常超过10年。新的药物开发模式引入了“注册导向的一期试验”,在这类试验中,扩展队列不仅关注安全性,还在具有充分统计动力的特定疾病队列中探索药物的活性。此外,多个联合疗法队列可以作为初期一期试验的一部分,并在适当的方案修订后进行,以评估不同药物组合的安全性和初步疗效。这些并行的多队列试验可能涵盖数百名患者,可能导致药物获得突破性指定、条件批准或加速批准,从而显著缩短整体的药物开发时间。特定患者队列也可以开放,以便评估药物在存在特定合并症的患者群中的安全性。同时,其他患者可以加入已证明安全且高于最小活性剂量的剂量递增阶段。这允许在多个剂量水平上丰富安全性、药代动力学(PK)、药效学(PD)及疗效数据,增加可能从药物治疗中受益的患者数量,而不会延误剂量递增过程。这里的关键术语包括最小有效剂量(MAD)、最大可行剂量(MFD)、最大耐受剂量(MTD)、推荐二期剂量(RP2D)。
32.2.2 安全性与毒性管理
本书中有一整章详细讨论了免疫肿瘤学(I-O)的副作用,这里我们将概述一期临床试验中的关键概念和历史进展。
在一期临床试验中,需要特别关注免疫毒性(免疫相关不良事件,irAEs)的几个特点(Fig. 32.3)。首先,irAEs可能影响任何器官,虽然抗CTLA-4和抗PD-1/PD-L1药物的早期试验主要关注结肠炎和肺炎,但实际上,任何器官的irAEs都应在病历中被仔细且详尽地记录。其次,irAEs虽然发生率不高,但可能迅速恶化并带来生命危险,因此必须教育患者及时报告任何症状,避免延误或自行处理。第三,鉴于irAEs的特殊管理需求,一期试验方案中应包含明确的毒性管理指南,并要求一旦irAEs达到2级,患者应立即由相关器官的专家进行处理。理想情况下,一期试验单位应与器官专家建立合作网络,以优化irAEs的识别和管理。这不仅有助于优化治疗,还可以在应用任何免疫抑制剂之前,从经历毒性反应的患者那里收集组织或血液样本,进行生理病理研究。
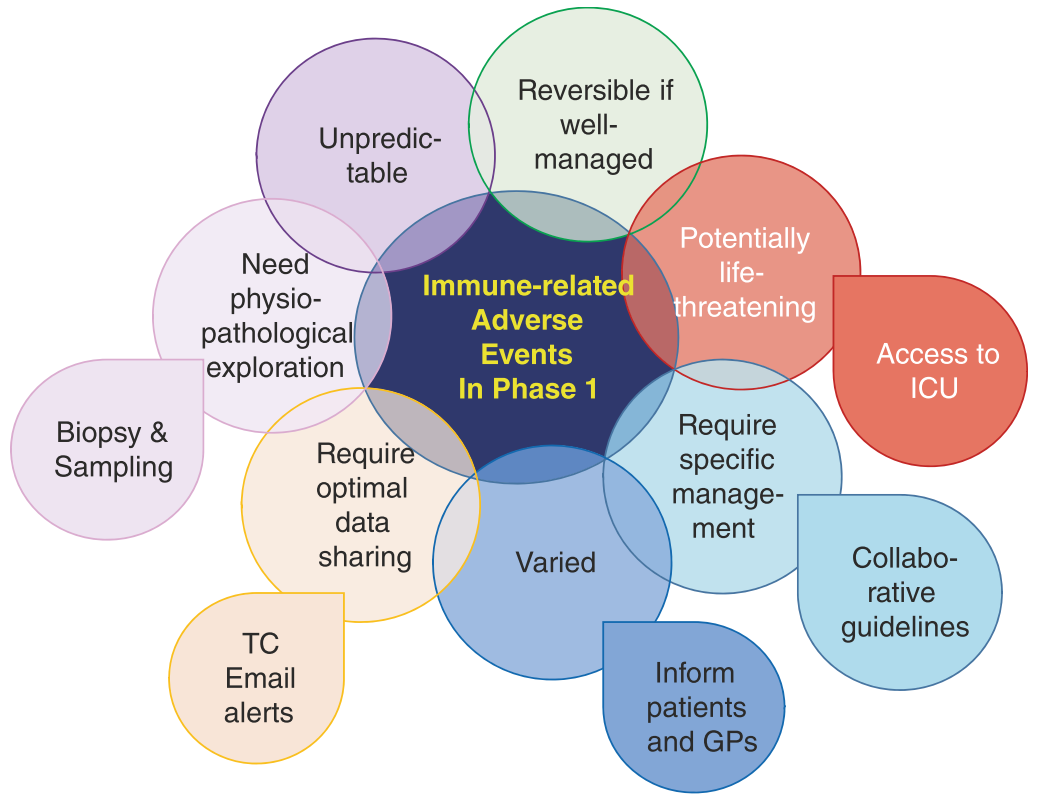
Fig.32.3 在一期试验中,针对免疫相关不良事件(irAEs)的特殊性需要特别关注。这些不良事件可能是多变且不可预知的,但如果管理得当,往往是可逆的。关键在于进行深入的生理病理学探索,包括必要时进行活组织检查和取样。irAEs可能具有潜在的生命威胁性,因此在出现严重事件时,确保能迅速进入重症监护室(ICU)至关重要。此外,这些不良事件的管理需要特定的措施,并依赖于全科医生(GPs)、重症监护专家和相关医疗团队的紧密合作。为了有效管理和快速响应,建立优化的数据共享系统非常重要,这包括电话会议(TC)和邮件提醒系统,以确保所有关键信息及时传达。此外,应制定协作性的治疗指南,明确每一步的管理策略,包括在不同情况下的具体干预措施。通知和教育患者及其全科医生关于可能遇到的irAEs及其变化性是必要的。通过这种方式,可以提升患者及医疗提供者对这些事件的认识和应对能力,从而有效地降低风险并优化治疗成果。
此外,有关严重毒性事件的历史案例提供了重要的教训,例如一种针对免疫突触的治疗方法,由于起始剂量选择不当而导致的严重不良反应。这个事件说明,起始剂量应基于最小生物效应水平(MABEL)而非传统的未观察到不良反应水平(NOAEL),并需考虑人类与动物(包括小鼠和猕猴)免疫系统间的差异。这种严重的毒性反应使得相关治疗方法的发展数年处于停滞。这一案例,连同其他疫苗和CAR-T细胞治疗的讨论,凸显了I-O药物毒性的不可预测性,它们虽然罕见但通常严重,并需要在专业单位进行及时且具体的管理。因此,对于首创或首次用于人体的药物,免疫疗法的一期试验至少应在具备丰富经验的大型一期中心进行,这些中心能够立即提供重症监护和有效的免疫抑制治疗(包括抗TNF和抗IL-6药物)。
32.2.3 药代动力学和药效学评价
32.2.3.1 药代动力学
考虑到免疫疗法的作用机制及当前阶段试验中涉及的药物种类,药代动力学评估正变得日益复杂。例如,大部分免疫检查点阻滞剂共有一个IgG骨架,这决定了它们剂量依赖的最大血药浓度(Cmax)和大约15天的中位半衰期,但根据不同的亚型,它们的作用机制存在显著差异。IgG1和IgG3可通过诱导自然杀伤(NK)细胞的抗体依赖性细胞介导的细胞毒性(ADCC)发挥作用,而IgG4则激活补体替代途径。此外,因素如浓度依赖的半衰期、Fc区域与新生抗体受体(FcRn)的结合能力,以及配体的循环可溶形式的存在,增加了这些药物药代动力学特征的可变性和复杂性。
与此相比,缺乏Fc区域的双特异性抗体通常具有非常短的半衰期,这对于管理药物不良反应(例如细胞因子释放综合征或抗药物抗体的出现)尤为重要。例如,双特异性T细胞接合(BiTE)抗体blinatumomab(Blincyto),针对CD19和CD3,其半衰期约为2小时。
最后,新的给药途径,如肿瘤内给药,带来了新的挑战。这要求不仅评估药物在外周血中的药代动力学,还要评估其在局部的效果。因此,对药物制剂的优化变得越来越重要,以确保药物在治疗区域的最大可用量和理想的药效。
32.2.3.2 药效学
设计适当的药效学分析方案,以评估免疫检查点阻滞剂的疗效存在困难,原因在于它们表达模式的高度变异性。例如,PD-1主要表达在T细胞表面,而CTLA-4通常仅为瞬时表达,且生物标志物的表达可能是固有的或是被诱导的。因此,利用流式细胞术来评估循环T细胞上的受体占用情况是可行的,但这种方法只适用于少数分子。这种技术已经成功应用于nivolumab的一期试验,研究发现受体占用率似乎与剂量无关,且持续时间较长(最后一次给药后3个月内观察到占用率下降)。
然而,仍然存在一些未解之谜,例如需要达到多大程度的目标调节和持续多长时间才能有效触发最佳免疫调节,以及在循环淋巴细胞中观察到的现象是否真实反映了肿瘤微环境中的情况。此外,研究强调了单克隆抗体Fc部分与先天免疫效应细胞上表达的Fcγ受体的相互作用在治疗活性中的重要性。已知携带激活型FcγR等位基因(具有更高亲和力的)的患者对治疗有更好的反应。例如,FcγRIIIα的多态性与滤泡性淋巴瘤患者对利妥昔单抗的反应密切相关。因此,已经开发了策略,通过限制或阻断抗体Fc部分与细胞Fcγ受体的结合来改变药物的疗效和药效学特性。
进行免疫监测的能力,特别是评估外周血中循环的细胞因子、趋化因子以及免疫细胞群的表型,为免疫治疗提供了一种有吸引力的药效学生物标志物。以9B12抗OX40单克隆抗体的一期试验为例,研究发现,药物输注后,患者的非调节性T细胞(FoxP3neg)CD4+ T细胞显著扩增,与此同时,表达CD38和HLA-DR激活标记的CD8+ T细胞也有所增加,而外周血中的滤泡辅助CD4+ T细胞则短暂减少。在拥有可供分析的肿瘤组织的三名患者中,通过比较发现,外周血中的调节性T细胞(Treg)OX40表达仅有不到20%,而在肿瘤浸润的Treg中,超过50%表达OX40。
此外,已有报道指出抗CTLA-4治疗可以剂量依赖性地增加FoxP3+ 和FoxP3- CD4+ T细胞。这种广泛而全面的药效学研究在一期临床试验中至关重要,尽管这些研究可能无法证明生物标志物在统计学上具有显著差异。实际上,一期试验是评估不同剂量计划和药物暴露对药效学(PD)参数影响的唯一机会。此外,这种研究还能够鉴定关键生物标记物,这些标记物在后期试验中可以优先评估,并招募更多患者,从而在一定程度上降低这类分析的成本。
32.2.4 病人资格
在当前的免疫治疗领域,特别是考虑到免疫治疗相关的特殊生理病理特征及大多数制药公司采用的“一期疗效和注册试验”策略(Fig 32.2),人们开始质疑是否应该重新考虑传统的一期临床试验入选标准。通常,一期试验的患者需满足以下条件:(1)预期寿命超过18周;(2)无重大基线器官功能障碍。传统上,预期寿命的评估采用化疗患者的客观评分系统,如基于LDH、白蛋白和转移位点数量的皇家马斯登评分。最近的研究重新审视了这种评分系统与免疫治疗患者的相关性,并提出了将淋巴细胞计数纳入评分系统的可能性。
然而,对于评估免疫疗法的一期试验而言,排除任何有基线器官功能障碍的患者似乎形成了一个悖论。实际上,最新的数据表明,针对免疫检查点的抗体治疗相关的毒性可能比化疗或靶向药物的毒性更为罕见且更多样。尽管如此,一期临床试验的纳入标准变得更加严格。从逻辑上讲,有自身免疫病(如牛皮癣、1型糖尿病、炎症性肠病等)、慢性病毒感染(如乙型肝炎、丙型肝炎、HIV)或有严重过敏或超敏反应史的患者通常被排除在外。
然而,如果在新药的初期开发阶段,即剂量递增阶段或首次在人体内测试的化合物阶段,这种严格的入选标准是合理的,那么它进一步降低了一期试验患者的代表性。特别是在制药公司计划根据一期试验结果加速某些化合物的批准的情况下,这种做法似乎违背了直觉。例如,在接受tremelimumab治疗的LDH升高的患者中,以及在使用BMS-936559治疗的两例患者中,已观察到反应和临床益处,包括那些患有脑转移的患者而未出现额外毒性的案例。这些发现提示我们在设定一期试验标准时可能需要更加灵活和包容。
32.2.5 患者选择和个性化免疫治疗
免疫治疗为患者选择和个性化医疗带来了额外的复杂性。与传统的癌症治疗相比,生物标志物不仅存在于癌细胞或由癌细胞释放的分子上,还需要关注免疫细胞及肿瘤微环境。此外,一些生物标志物如PD-1或PD-L1,表达具有动态变化性,可能仅在特定条件下被诱导表达。选择预测免疫治疗疗效的最佳生物标志物的挑战——例如检查点表达、新抗原/突变负荷、免疫评分等——虽已在本书的前一章进行讨论,但一期临床试验的特定需求仍需强调。
首先,一期试验需要在时间上尽可能高效,并能够有效招募患者。因此,在剂量递增阶段不应进行基于分子的选择,其主要目标是确定剂量限制毒性和最大耐受剂量。随后,在专用的生物标志物队列中,可以在剂量扩张阶段进行分子富集。例如,22C3 IHC PD-L1 PharmDx测试的成功开发和批准,以及派姆单抗的使用,很好地体现了这一点。
其次,当前用于预测免疫疗法反应的生物标志物,尤其是免疫检查点阻滞剂,普遍缺乏足够的特异性或敏感性来确定哪些患者不应接受此类疗法。例如,在PD-L1阴性的人群中或在几乎没有突变且新抗原负荷极低的肿瘤类型(如霍奇金淋巴瘤)中,仍然观察到了对抗PD-1或PD-L1药物的反应。如果在一期试验中过早地采用严格的预选标准,可能会错过这些重要的观察结果。
第三,有时“意外”反应者的出现可以为我们提供比精心挑选的同质患者队列更多的信息,帮助我们更深入地了解药物的作用机制和潜在的目标人群。这种发现强调了在一期试验中保持入选标准的灵活性的重要性。
一期临床试验提供了一个独特的机会,因为其入选标准足够灵活,可以招募各种患者,包括罕见肿瘤类型或未特定选择的患者群体。因此,在剂量扩展阶段,应系统地纳入这些特殊患者队列。此外,对于那些表现出异常应答或耐药性的患者,应在肿瘤组织、外周血,以及可能的情况下,在淋巴结中进行广泛的取样和研究,因为这些患者可能提供极具价值的信息。
值得关注的是,在过去三年中,追求相同治疗目标的公司数量不断增加,从26%增加到36%,而只有一家公司追求的目标数量从42个减少到26个。尽管目前还没有具体的肿瘤免疫治疗数据,但观察到大多数免疫肿瘤药物公司都在沿用相同的路径,不仅开发针对同一分子的药物,还针对相同的患者群体。这种趋同现象不仅使得一期临床试验的招募变得更加困难,而且由于试验之间的竞争,也在不断减少针对特定患者群体(如肉瘤、脑瘤或罕见疾病)进行新药开发的可能性,进而损害了真正的创新。这种情况强调了为特殊患者群体设计独特治疗方案的重要性,以及在一期试验设计中采纳灵活多样的策略的必要性。
本文由Oncolab微信公众号翻译整理自Oncoimmunology: A Practical Guide for Cancer Immunotherapy,仅用于学习交流使用,如需购买原版书籍可点击阅读原文购买。
— THE END —
来源 | Oncoimmunology: A Practical Guide
for Cancer Immunotherapy
初稿翻译 | 冼金欢
图文排版 | 潘君君
审核修改 | 王坤
(转载请保留此部分内容)

关注本号~

加入读者交流群~
(添加请备注单位姓名)

加入知识星球~
点亮赞与在看
让更多人看到
本篇文章来源于微信公众号: OncoLab
 微信扫一扫打赏
微信扫一扫打赏
 支付宝扫一扫打赏
支付宝扫一扫打赏