讨论交流及PDF下载可移步OncoLab学术星球~
摘要
在癌症发生和发展过程中存在免疫系统和肿瘤细胞之间复杂的相互作用。虽然免疫系统在早期消除了恶性转化的细胞,但存活的肿瘤细胞仍可以通过各种方法逃脱宿主的免疫防御,甚至重新编程对前肿瘤表型的抗肿瘤免疫反应,以获得无限的生长和转移。肿瘤细胞的高增殖率增加了肿瘤细胞对局部营养物质和氧气的需求,而不良的组织血管几乎不能满足这一要求,这就会造成酸性、缺氧和葡萄糖缺乏的肿瘤微环境。因此,肿瘤微环境中的脂质被激活,并作为肿瘤细胞和相关免疫细胞的主要能量来源和关键调节因子被利用。然而,脂质代谢重编程在肿瘤免疫反应中的确切作用尚不清楚。全面了解肿瘤微环境中的脂质代谢功能障碍及其对免疫反应的双重影响,对于绘制肿瘤免疫学的详细图景和开发癌症患者的特定治疗方法至关重要。在这篇综述中,我们集中讨论了肿瘤微环境中脂质代谢的失调,并讨论了它在肿瘤免疫应答中的相互矛盾的作用。此外,我们总结了目前肿瘤免疫治疗中针对脂质代谢的治疗策略。本综述总结了脂质代谢在肿瘤免疫应答中的作用。
关键词
脂质代谢、恶性肿瘤、免疫反应、肿瘤微环境、免疫治疗。
背景
免疫系统的关键功能是通过识别独特的抗原来区分转化细胞和正常细胞,并通过体液和细胞介导的免疫反应对抗恶性肿瘤。许多内在的分子在癌症开始的早期阶段就会发生变化,导致机体产生独特的免疫反应应答。肿瘤细胞产生的一些因子,如双链DNA,可触发宿主免疫系统产生有效的免疫应答,降低肿瘤的发生率。这种抗肿瘤免疫反应也被称为“免疫监视”,在恶性肿瘤的整个发展过程中通常发挥着消除和控制癌细胞的作用,特别是在癌症早期阶段。然而,免疫系统并不是总能发现并杀死所有的癌细胞。这些存活的肿瘤细胞会重塑局部免疫细胞和基质细胞,为自己建立一个称为“肿瘤微环境(TME)”的生存环境,以逃避免疫监测和消除。
TME中的许多因子驱动免疫细胞向肿瘤前表型转变,从而促进肿瘤细胞的增殖和转移。而这些免疫反应是基于肿瘤浸润免疫细胞产生的,这些肿瘤浸润免疫细胞包括T细胞、巨噬细胞、树突状细胞(DCs)、中性粒细胞(Ns)、自然杀伤细胞(NK)和骨髓源性抑制细胞(MDSCs)。
T细胞是整个肿瘤发展过程中的关键参与者。CD8+ T细胞可分化为细胞毒性T淋巴细胞(CTL),并以主要组织相容性复合体(MHC)依赖的方式直接清除肿瘤细胞。CD4+ T细胞包括促炎性T辅助性1(T1)细胞、免疫抑制T辅助性2 (T2)细胞、模糊T辅助性17 (T17)细胞和免疫调节性T细胞(Tregs)。每一种都在肿瘤免疫发挥独特的作用。
肿瘤浸润巨噬细胞可以分为经典活化(M1)巨噬细胞和交替活化(M2)巨噬细胞。M1巨噬细胞容易通过产生促炎细胞因子和诱导型一氧化氮合酶(nitric oxide synase, iNOS)参与抗肿瘤免疫应答。相反,M2巨噬细胞表现出抗炎表型,会分泌许多促肿瘤因子,如arginase 1 (ARG1)。
虽然Ns是参与炎症过程的重要免疫细胞,但它们以两种不同的方式影响TME。N1亚型分泌促炎细胞因子,引起细胞毒性免疫反应,N2亚型被认为是免疫抑制细胞。
MDSCs是一组不成熟的髓细胞,在抑制肿瘤的发展、转移和治疗中发挥重要作用。根据其来源、组织定位和免疫抑制机制,MDSCs可分为单核细胞MDSCs和多形核MDSCs。它们通过抑制其他免疫细胞的方式表现出免疫抑制能力,而该能力与其所在环境有关。
此外,NK细胞和DC被认为在抗肿瘤免疫应答中也发挥了重要的作用,但各种免疫抑制因子往往会抑制其能力,使其成为促肿瘤表型。
免疫细胞外,癌症相关成纤维细胞(CAFs)是肿瘤间质的主要成分,对TME的形成有重要作用。CAFs可通过沉积、重塑细胞外基质、分泌细胞因子或与细胞直接接触,促进肿瘤细胞向恶性表型转化,增强肿瘤细胞逃避免疫检测和消除的能力,调节其他肿瘤浸润免疫细胞。
在细胞生存中能量和代谢产物是必不可少的。恶性肿瘤细胞需要大量的能量和原料来支持其不受控制的增殖和对子细胞的维持。虽然TME内血管生成增加,但葡萄糖和氧气的量仅能满足癌细胞扩张和扩散的需求,因此会导致葡萄糖缺乏和缺氧的微环境。这会迫使恶性肿瘤细胞调整其代谢谱以维持自身的增殖。
20世纪20年代,Otto Warburg等人首次提出,无论TME中含氧量如何,肿瘤组织在给定时间内代谢葡萄糖转化成乳酸的量大约是正常组织的10倍,这被称为Warburg效应。此后,代谢重编程成为恶性肿瘤研究中一个十分具有研究前景的领域(Fig. 1)。
近年来,脂质对肿瘤发展的影响越来越受到人们的关注。脂质除了被用作机体能量短缺时的替代能源外,还参与生物膜的合成,为生物质生产提供基质,并激活与癌细胞生长和迁移相关的复杂信号通路。癌细胞会表现出脂质和胆固醇需求增加的现象,如外源性脂质和脂蛋白摄入增加,及脂质和胆固醇的从头合成过度激活,都会直接导致肿瘤细胞的恶性转化和转化进展,及TME中的脂质异常积累(Fig. 1)。
TME中的脂质代谢障碍对肿瘤相关免疫反应具有深远的影响。根据对肿瘤细胞的作用,免疫应答可分为促肿瘤免疫应答和抗肿瘤免疫应答,两者均涉及多种免疫细胞,如T细胞、巨噬细胞、DCs、MDSCs等。由于脂质的复杂组成和潜在的机制,同一类型的免疫细胞可能对脂质代谢的变化作出非常不同的反应,从而得使研究得出模棱两可的结论。
例如,过量的游离脂肪酸(FFAs)抑制CTL介导的对肿瘤细胞的杀伤,可以通过降低FFA水平来恢复。然而,在严酷的TME中,组织驻留记忆(Trm)细胞倾向于通过CD36吸收FAs来支持抗肿瘤反应和维持它们自身的长期生存。因此,确定免疫细胞脂质代谢的确切机制对于理解免疫抑制现象和开发有效的联合疗法至关重要。
在本文中,我们从两个角度讨论了脂质代谢对肿瘤相关免疫反应的复杂作用。此外,我们还探讨了目前针对TME中脂质代谢的策略和分子靶点,并描述了其局限性和未来的发展方向。
脂质代谢的基本概念
脂质根据其结构可分为三大类:(a)含有长链FAs及其酯化产物的简单脂质,如三酰基甘油(TAG); (b)衍生脂质,如类固醇、脂溶性维生素和类胡萝卜素;(c)含有各种官能团的复合脂质,如磷脂、糖脂质和脂蛋白(LP)。
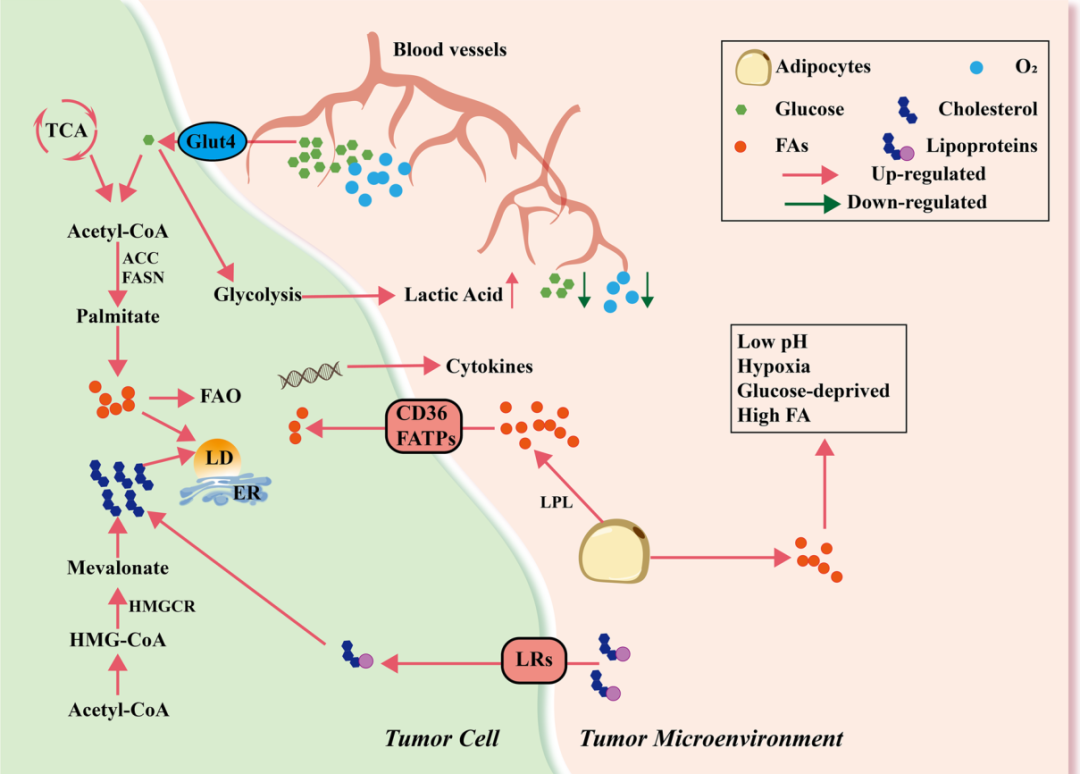
Fig. 1 肿瘤微环境中的代谢重编程。在肿瘤微环境中,紊乱的血管运输的葡萄糖和氧气大部分被肿瘤细胞吸收,导致缺氧和缺糖的微环境。肿瘤细胞中激活的糖酵解能产生更多的乳酸,但仍不能满足能量需求。因此,来自肿瘤细胞和其他基质细胞的LPL(脂蛋白脂肪酶)激活脂肪细胞,诱导储存的甘油三酯的脂解和FAs(脂肪酸)的分泌,并通过CD36或FATPs(脂肪酸转运蛋白)转运到细胞内。此外,肿瘤细胞可以通过葡萄糖分解代谢,利用乙酰辅酶a通过从头合成途径生成FAs(脂肪酸)。ACC(乙酰辅酶A羧化酶)和FASN(脂肪酸合成酶)参与这一过程。这些FAs随后参与FAO(脂肪酸氧化)或其他信号通路,产生许多免疫抑制因子或产生LDs(脂滴)。此外,TME中的脂蛋白通过脂蛋白受体(lipoprotein receptor, LRs)运输,并在细胞中分解为胆固醇。肿瘤细胞也可以通过甲戊酸途径产生胆固醇。肿瘤细胞的脂质代谢异常促进了酸性、缺氧、缺糖和高脂免疫抑制微环境的形成。
脂肪酸
外源性FAs主要来源于TAGs, TAGs主要在空肠上端被胰脂肪酶催化消化。这些FFAs通过CD36或脂肪酸转运蛋白(FATP)从肠腔被吸收到肠上皮细胞,然后通过单酰基甘油酰基转移酶和二酰基甘油酰基转移酶重新组装成TAG。为了在血液中运输,TAGs通常与脂多糖结合,形成乳糜微粒、极低密度脂多糖(VLDL)、低密度脂多糖(LDL)和高密度脂多糖(HDL)。脂多糖可被脂蛋白脂肪酶(LPL)水解或通过相应受体转运到细胞内,生成FAs和蛋白。
从头合成是FAs的另一个主要来源。ATP -柠檬酸裂解酶(ACLY)催化柠檬酸转化为乙酰辅酶A (acetyl-CoA),然后乙酰辅酶A羧化酶(acetyl-CoA carboxylase, ACC)将乙酰辅酶A转化为丙二酰辅酶A。脂肪酸合成酶(FASN)通过乙酰辅酶a和丙二酰辅酶a的反复缩合生成棕榈酸,棕榈酸通过进一步伸长和去饱和作用转化为许多其他类型的FAs(Table 1)。
在细胞中,FAs主要通过脂肪酸氧化(FAO)作为一种能量来源。脂肪酰基辅酶A合成酶(ACS)和肉碱棕榈酰转移酶I (CPT1)是生成脂肪酰基辅酶A和脂肪酰基肉碱的关键酶。酰基肉碱进入线粒体后,转化为酰基辅酶a,进入三羧酸循环生成三磷酸腺苷 (ATP)和烟酰胺腺嘌呤二核苷酸磷酸(NADP),从而抵消氧化应激。
此外,FAs通过结合和激活核受体家族的转录因子,如过通过氧化物酶体增殖物激活受体(PPARS)来调节细胞信号通路和基因转录,PPARS控制脂质、能量稳态和炎症相关基因的表达。PPAR-α直接上调CPT1α和PPAR-δ的表达,调节脂质输送和氧化。
此外,固醇调节元件结合蛋白1 (SREBP-1)转录因子影响参与脂代谢的基因如ACLY、ACC和FASN[30]的表达(Table 1)。
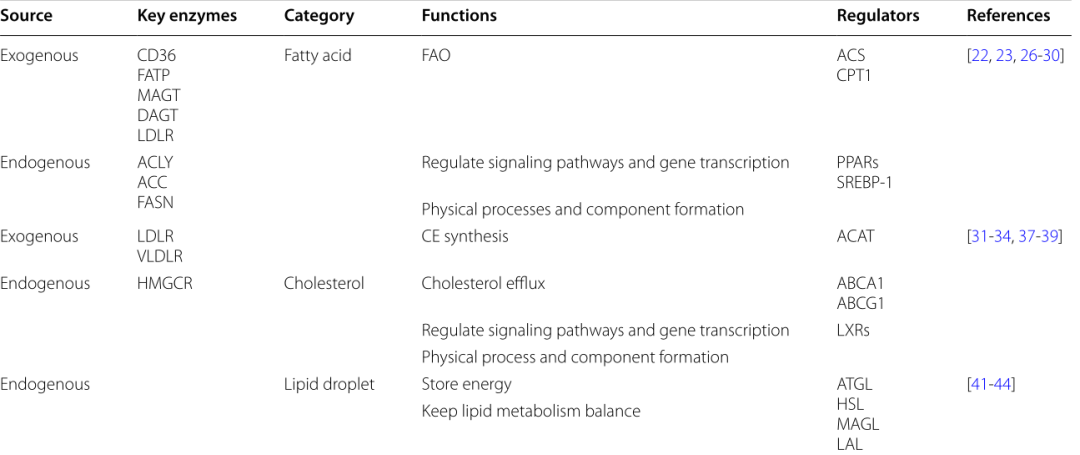
Table 1 初级脂质的基本概念
胆固醇
胆固醇是脂质代谢不可缺少的组成部分。它的吸收伴随着FAs通过CD36和尼曼- pick C1 like 1,然后它们重新组装成乳糜微粒。与FAs类似,肝脏中胆固醇被包装成非常低密度的脂多糖和低密度脂多糖,并在循环中运输。这些脂多糖通过相应的受体进入细胞后,被溶酶体中的胆固醇酯水解酶水解释放胆固醇。
然而,饮食来源的胆固醇不能满足生理需求,其通过甲戊酸途径的从头合成才是胆固醇的重要来源。3-羟基-3-甲基戊二酰基辅酶A还原酶(HMGCR)是该过程中的限速酶,介导3-羟基-3-甲基戊二酰基辅酶A转化为甲戊酸,甲戊二酰基辅酶A进入下游进行多步合成(Table 1)。
为了避免游离细胞间胆固醇大量积累而产生细胞毒性,酰基辅酶a -胆固醇酰基转移酶(ACAT)将胆固醇转化为胆固醇酯(CE),并将其储存在脂滴中(ID)。ACAT2缺乏可显著降低胆固醇的吸收速率,激活SREBP,增强HMGCR、脂蛋白受体(LDLR)等许多调节蛋白的转录,增加胆固醇的摄取和合成。此外,ATP结合盒(ABC)蛋白会介导细胞内胆固醇外流,尤其是巨噬细胞。
肝脏X受体(Liver X receptor, LXR)促进脂肪生成和ABCA1、ABCG1的转录。许多物理过程依赖于胆固醇,如质膜、小泡和脂质的形成,一些如dolichoL和辅酶Q的分子的生物合成、以及代谢物的结构骨干形成。然而,过多的胆固醇会降低膜流动性,破坏脂质信号,产生破坏性的氧化分子,最终导致细胞死亡(Table 1)。
脂滴
LDs是一种重要的亚细胞器,通过维持脂质储存和分解的平衡来调节细胞脂质代谢,并保护细胞免受潜在的有毒脂质的侵害。LD包含一个由TAG、固醇酯和FFAs组成的脂质核,它被一个由磷脂和胆固醇组成的单层膜包围。
脂质代谢中涉及的各种酶和蛋白,如脂肪甘油三酯脂肪酶(ATGL)、激素敏感脂肪酶(HSL)、单酰基甘油脂肪酶(MAGL)、溶酶体酸性脂肪酶(LAL)等都位于LDs表面,参与脂质代谢的调节。PPAR激动剂、糖皮质激素和禁食会提高ATGL表达,而胰岛素和食物摄入则有相反的作用(Table 1)。
在TME中,这些复杂的脂质成分及其代谢过程受到异常调节,从而对肿瘤免疫应答产生重要影响。
脂质在抗肿瘤免疫应答中的作用
TME中血管交换不足导致的营养限制可导致免疫细胞与快速增殖的癌细胞之间的激烈竞争,从而改变抗肿瘤免疫防御功能。免疫细胞和免疫介质被激活参与宿主对恶性肿瘤的防御。CTLs、NK细胞、巨噬细胞和DCs是肿瘤检测和清除的主要效应细胞。尽管肿瘤细胞会形成一种严酷的TME,但这些效应免疫细胞仍会找到新的生存方式,并使用脂质作为支持抗肿瘤免疫反应的燃料。
脂质在抗原特异性抗肿瘤免疫反应中的作用
CD8+ T细胞被认为是适应性抗肿瘤免疫中最重要的执行者。肿瘤特异性MHC限制性CTLs可以在不影响正常细胞的情况下溶解肿瘤细胞。
FAs作为T细胞的重要组成部分,它可以通过FAO和磷酸化为T细胞提供能量。TME中增加的FAs激活CD8+ T细胞中的PPARα信号,促进其脂质代谢并维持效应功能。
此外,促进肿瘤膨胀型CD8+ T细胞的FA分解代谢可提高其抗肿瘤活性。PPAR-γ是脂质代谢中的一种关键的配体诱导核受体,激活PPAR-γ可以通过上调FAO和抑制Tefs的凋亡,来促进天然T细胞向效应T细胞(Tefs)的分化。并且,CD8+ T细胞增加了引流淋巴结和肿瘤部位中效应/记忆性CD8+ T细胞的数量和活性(Fig. 2a)。
CD8+ T细胞中脂质的消耗能够阻碍其增殖和细胞毒性,导致功能衰竭,其特征是干扰素(IFN)-γ和高度程序性细胞死亡蛋白-1 (PD-1)表达减少。PD-1信号通路抑制TCR-和CD28介导的PI3K/AKT/ mTOR通路的激活,进而促进脂肪分解和FAO。
有报道称,抗PD -1抗体的应用抑制了T细胞的FAO水平,并导致T细胞过度激活而导致细胞死亡。这些结果表明,将FAO激活因子与抗PD -1抗体结合可以逆转FAO水平的抑制及其对T细胞分化的抑制作用。
一种pan-PPAR受体激动剂Bezaf brate可提高PD-1阻断下的FAO水平,提高CD8+ T细胞的增殖和抗肿瘤能力。相比之下,PPAR-α基因敲除会损害伴有PD-1表达的CD8+ T细胞的FA分解代谢和抗肿瘤活性。
除Tefs外,肿瘤膨胀型CD8+ Trm T细胞还通过释放溶解颗粒、颗粒酶B (GzmB)、IFN-γ和肿瘤坏死因子(TNF)-α表现出抗肿瘤活性(Fig. 2a)。CD36介导的细胞间高FA水平有助于维持CD8+ Trm细胞的有效功能和长期生存,逆转由癌细胞引起的脂质剥夺所致的细胞凋亡。
此外,程序性死亡配体1 (programmed death-ligand 1, PD-L1)堵塞可增加4/5的脂肪酸结合蛋白(FABP) 的表达和对脂质摄取,延长Trm细胞在体外和体内的生存期。
这些结果提示FA代谢和T细胞抗肿瘤作用之间可能存在的潜在联系,特别是免疫检查点方面。增强CD8+ T细胞中的FAO可以增强其抗肿瘤反应,尤其是与抗PD -1治疗联合使用时。因此,FA代谢至少部分增强了T细胞介导的抗肿瘤免疫反应,这意味着刺激T细胞FA摄取和增强FAO可能是一种可以运用于治疗恶性肿瘤的方法。所以,恶性肿瘤的临床治疗应结合T细胞脂质代谢的干预。
为了验证这一假设,研究人员已经探索了FAs在嵌合抗原受体(CAR) T细胞中的作用,这是治疗复发或难治性恶性肿瘤的一种有效选择。
4-1BB CAR-T细胞促进CD8+中央记忆T细胞的生长,该细胞以FAs为主要能量来源,广泛依赖于FAO。与CD28 CAR- T细胞相比,CAR架构中的4-1BB导致了更高的FAO、呼吸能力和生长速率,CD28 CAR- T细胞的特征是高糖酵解。这些结果表明,CAR-T细胞中FA代谢的改变可以影响它们的生存、功能和注射后的抗肿瘤免疫反应,这为我们提示了一个很有前景的T细胞治疗方向。
胆固醇是T细胞介导的抗肿瘤免疫反应的另一个关键调节因子。T细胞膜中的游离胆固醇是T细胞受体(TCRs)和T细胞免疫突触的重要组成部分,直接调节信号通路和效应器功能(Fig. 2a)。ACAT1/2抑制剂阻断胆固醇酯化作用导致胆固醇水平升高,促进TCR微簇直接向突触中心移动,同时CD8+ T细胞数量和抗肿瘤活性均升高。同时,在ACAT1抑制CD19-CAR-T细胞处于低感染率的情况下,通过提高效应靶细胞比例,增强了CD19-CAR-T细胞的限瘤活性,并可在化疗免疫治疗中拮抗CD8+ T细胞抑制。
此外,自然杀伤T细胞(natural killer T, NKT)细胞是一群CD1d限制性T细胞,同时具有适应性和先天免疫细胞的特征,主要功能是识别脂质抗原,发挥多种免疫调节作用。因此,脂质可以调节NKT细胞的细胞毒性。肿瘤细胞的过度糖酵解导致TME中产生大量的乳酸,从而降低肿瘤膨胀不变NKT细胞(iNKT)中PPAR-γ的表达水平,进而减少脂质合成和IFN-γ的产生。PPAR-γ激活剂逆转了这一现象,增强了iNKT的抗肿瘤活性(Fig. 2c)。
此外,体外实验表明,只有水溶性胆固醇,而不是其他类型的脂质,能够成功恢复IFN-γ的产生,并增强iNKT细胞免疫突触的TCR信号。辛伐他汀抑制胆固醇合成显著削弱了这种作用。
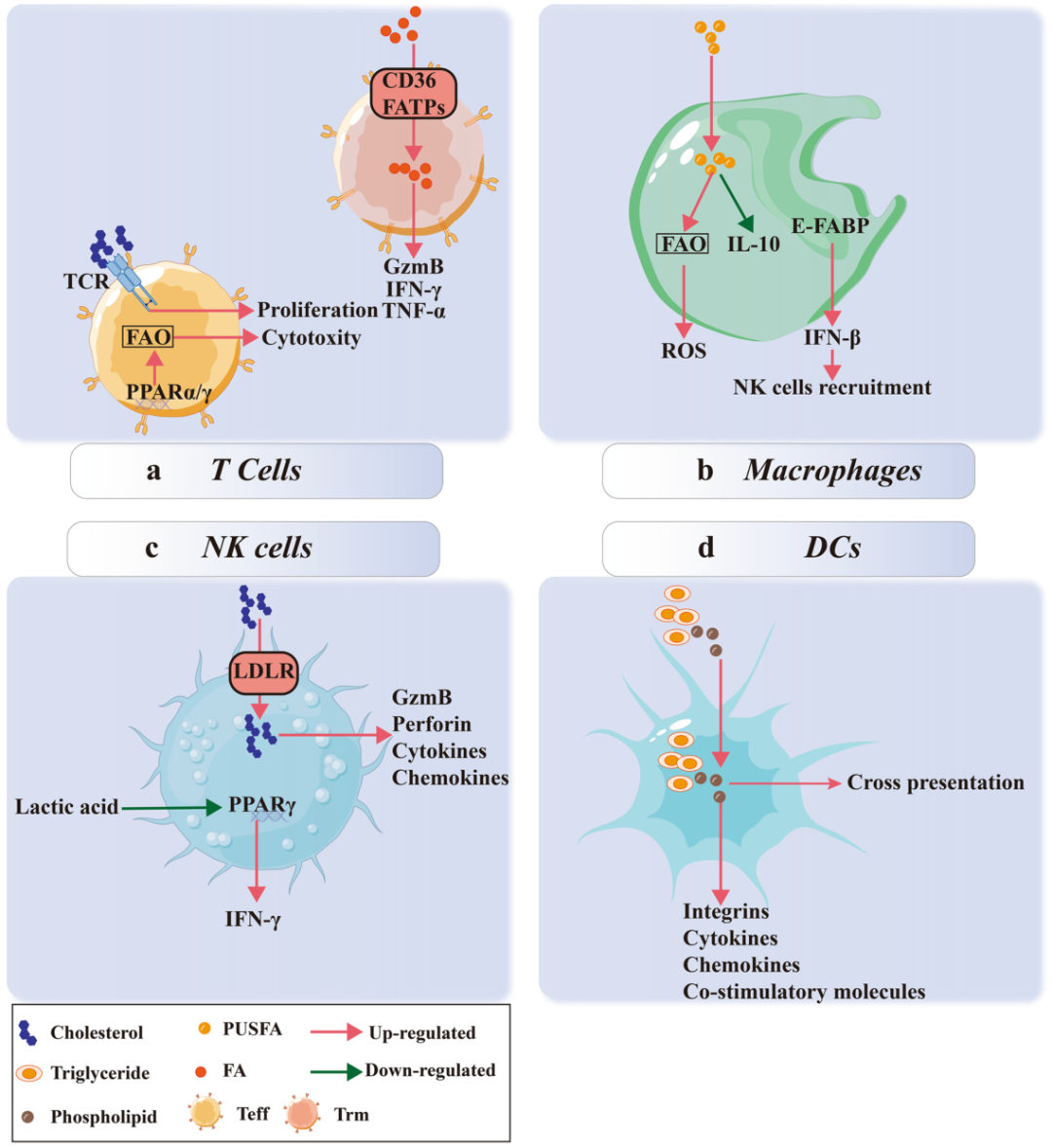
Fig. 2 抗肿瘤免疫反应中的脂质代谢。Trms通过CD36和FATPs从TME中吸收FAs,产生抗肿瘤细胞因子如GzmB, IFN-γ和TNF-α。此外,胆固醇有助于Tefs上tcr的形成,并刺激其增殖和细胞毒性。PPAR-α/γ也可通过激活FAO增强Tefs的抗肿瘤能力;b TME中的FAs增强巨噬细胞中的FAO,从而上调ROS的产生,下调IL-10的分泌,消除肿瘤细胞。巨噬细胞E-FABPs水平升高可促进IFN-β表达,介导NK细胞招募杀伤肿瘤细胞;c通过LDLR摄入的胆固醇刺激效应标记物(GzmB和穿孔素)、细胞因子和趋化因子的表达。NK细胞激活PPAR-γ促进IFN-γ分泌,TME中过量乳酸抑制IFN-γ分泌;d甘油三酯和磷脂增强树突状细胞的交叉表达能力和细胞因子分泌水平,参与抗肿瘤反应。
脂质促进非特异性抗肿瘤免疫反应
FAs和胆固醇作为肿瘤浸润T细胞的燃料和结构成分,促进抗原特异性检测和肿瘤细胞的清除。与NKT细胞不同,NK细胞诱导的抗肿瘤免疫反应是非特异性的。
NK细胞主要的抗肿瘤机制包括效应物对靶点的识别和偶联、死亡信号的传递以及肿瘤细胞的崩解。胆固醇能增强NK细胞的抗肿瘤活性。高胆固醇饮食可增加NK细胞的数量,并上调NK细胞中激活受体和效应蛋白的水平,如GzmB和穿孔素。LDLR介导NK细胞中胆固醇的积累,会刺激免疫激活能力,降低肿瘤增殖(Fig. 2c)。在肿瘤浸润的NK细胞中特异性表达LDLR可能是动员NK细胞产生抗肿瘤免疫反应的最佳方法。
因此,胆固醇在NK细胞中的积累可能是一种高效的方法,可以在免疫抑制TME中唤醒它们的抗肿瘤能力,并单独或协同发挥肿瘤消除作用。
在TME中,巨噬细胞和树突状细胞主要通过抗体依赖的细胞毒性和可溶性细胞毒性因子发挥抗原呈递作用,并发挥抗肿瘤作用。有报道称,肥胖会积聚促炎巨噬细胞,并导致活性氧(ROS)水平升高(Fig. 2b)。
然而,这种现象并不普遍,与FA亚型有很大关系。例如,与椰子油喂养的小鼠相比,鱼油喂养的小鼠肿瘤前巨噬细胞和白介素(IL-10)的表达比例较低,B细胞和CD+ 8 T细胞的浸润率较高。此外,多不饱和脂肪酸(PUSFAs)而非饱和脂肪酸(SFAs)的富集对巨噬细胞对肿瘤细胞具有更高的细胞毒性。
这就产生了一种假设,即一些FAs可能增强巨噬细胞的抗肿瘤免疫应答,而另一些则没有。鱼油中的一种不饱和脂肪酸N-3 docosapentaenoic acid特异性地增加了粮农组织相关基因(A-FABP、Cpt1b、PCX和UCP2)和凋亡相关基因(iNOS、RIPK3、caspase-8和caspase-11)的表达,从而诱导ROS介导的巨噬细胞凋亡,降低了促肿瘤活性。高表达E-FABP的巨噬细胞增加了IFN-β的产生,招募了更多的抗肿瘤免疫细胞,尤其是NK细胞(Fig. 2b)。
此外,酶引导的磷酸酪氨酸胆固醇的自组装通过诱导ROS的产生,破坏巨噬细胞中的游离分子,抑制卵巢癌细胞的生长,使前肿瘤巨噬细胞重新分化为抗肿瘤表型。这提出了一种新的方法,通过提供外源性修饰脂质,使肿瘤浸润的巨噬细胞极化甚至重新培养为抗肿瘤表型。
与其他免疫细胞不同,脂质类型(饱和与不饱和)较脂质的数量,在刺激树突状细胞处理抗原或释放抗肿瘤细胞因子中发挥着更为关键的作用。脂质含量较高的树突状细胞倾向于积累磷脂,而不是胆固醇和胆固醇酯。
与脂质含量低的树突状细胞相比,这些树突状细胞具有更高水平的整合素、共刺激分子、糖蛋白、促炎细胞因子和趋化因子。体内实验表明,较高的脂质含量使树突状细胞具有较强的交叉呈现能力,能有效激活NK和NKT细胞,增强内源性CTL。低脂含量更容易引起能量不足。
这些结果在免疫高脂质树突状细胞的小鼠中得到了证实,结果显示,在小鼠脾脏和肝脏中均出现了强劲的靶细胞裂解,并加速了OVA限制性CD8+ Tefs的增殖,延缓了肿瘤的发展,使病灶变小。
虽然对于DC中脂质的研究还很有限,但可以看出FAs可能作为促炎因子,以DC依赖的方式增强CTL的抗肿瘤活性,从而抑制肿瘤的生长。但是,这种现象是否普遍存在于肝脏以外的其他组织和癌症类型,还需要进一步研究。
高度增殖的肿瘤细胞引起葡萄糖缺乏和脂质丰富的TME,已被证明有助于肿瘤细胞的生长和传播,并导致免疫抑制环境。但是 ,需要指出的是,脂质也可以作为不可或缺的燃料和代谢成分,促进免疫细胞的抗肿瘤活性。
目前针对恶性肿瘤的脂质代谢靶向治疗多致力于单纯阻碍TME中的泛脂质代谢,效果有限。但如前所述,某些免疫细胞的特异性激活代谢途径可显著刺激抗肿瘤免疫反应,促进抗肿瘤治疗。
脂质在肿瘤微环境中的免疫抑制作用
虽然免疫系统致力于监视和消除转化细胞,但一些狡猾的肿瘤细胞会伪装自己,导致免疫逃逸。为了营造适宜的生长环境,肿瘤细胞对周围的基质细胞进行改造,将免疫细胞从抗肿瘤表型改造为旁观者表型,甚至是促肿瘤表型。
如前文所述,肿瘤细胞与免疫细胞对营养物质的激烈竞争,最终导致微环境缺糖缺氧,脂质过度堆积,脂质代谢活跃。尽管脂质代谢激活会引起免疫细胞上述对肿瘤的抑制作用,但异常的脂质状态也会抑制免疫细胞的抗肿瘤能力,甚至使免疫细胞重新分化为促肿瘤表型。
这种不一致可能是由于癌症类型或脂质种类的差异导致的。因此,深入探讨脂质代谢和肿瘤前免疫应答对于在不破坏抗肿瘤免疫应答的前提下逆转免疫抑制TME的机制至关重要。
FAs的积累破坏了效应T细胞的细胞毒性
最近的研究报告,原生T细胞的能量产生更多地依赖于线粒体呼吸和FAO,以尽量减少有毒代谢物对细胞的破坏作用。然而,为了维持较高的增殖速率和活性,Tefs倾向于利用糖酵解和有氧呼吸来满足ATP的需求。这意味着富含脂质的TME可能会抑制Tefs的抗肿瘤作用。
由于缺少关键酶,CD8+ T细胞无法分解积累的细胞间极长链FAs或将其储存在LDs中,因此会造成严重的脂肪毒性,进而导致T细胞衰竭。富含脂质的人乳腺癌组织释放大量FFA以抑制CTL介导的肿瘤抑制,这可以通过降低FFA水平来逆转(Fig. 3a)。介导FAs的能力降低会使CD8+ T细胞无法维持脂质稳态并降低其抗肿瘤能力。而Tefs细胞中积累的FAs增强了随后的分解代谢和FAO。
虽然可以推测FAO可以提高PD-1抗体治疗时CD8+ T细胞的抗肿瘤活性,但结果却与此相反。有报道称STAT3通过促进FAO和减少糖酵解来抑制CD8+ Tefs细胞。在T细胞中敲除Stat3或使用FAO抑制剂可以增强糖酵解和抗肿瘤功能,从而消除乳腺肿瘤。
有报道称,在这一过程中,是通过与瘦素受体或CD8+ T细胞的PD-1结合,刺激STAT3和CPT1B,抑制其糖酵解和抗肿瘤能力,而瘦素(leptin)是脂肪细胞分泌的一种脂肪因子(Fig. 3a)。
此外,PD-1的激活改变了预先激活的CD4+ T细胞的代谢程序,从糖酵解转化为FAO。这些机制可能解释了PD-1刺激的T细胞免疫耐受的原因。
Tc9细胞是高表达IL-9的CD8+ T细胞亚群,具有比Tc1细胞更强的抗肿瘤能力。芯片分析显示,Tc9细胞PPAR-α/类维生素a X受体α水平较低,胆固醇合成酶(Hmgcr和Sqle)表达较低,胆固醇流出酶(Abca和Abcg1)表达较高。胆甾醇的加入可抑制IL-9的表达,诱导Tc9细胞凋亡,导致抗肿瘤活性受损。
除了影响细胞因子的分泌,胆固醇还会导致肿瘤浸润性CD8+ T细胞的衰竭(Fig. 3a)。体外实验和临床样本显示,胆固醇可增加CD8+ T细胞的PD-1和2B4的表达,且呈剂量依赖性,与细胞凋亡率相关。而CD8+ T细胞中GzmB、IFN-γ和TNF-α的增殖率和产量均下降。这一现象的可能机制至少有部分可能是依赖于X-box结合蛋白1的升高和内质网应激。
调节性T细胞需要FAs来发挥免疫抑制作用
Tregs是一组肿瘤浸润性免疫抑制CD4+ T细胞,主要依赖于FAO而不是糖酵解。这些特征使它们能够在营养耗尽的TME中生存并表现出免疫抑制作用。
在缺氧TME中,缺氧诱导因子-1α引导葡萄糖远离线粒体,促进Tregs中的线粒体代谢,增强其抑制CD8+ T细胞的能力。叉头盒蛋白P3 (Forkhead box protein P3, FoxP3)可促进Tregs中的FA摄取、氧化磷酸化和FAO,并帮助它们在脂毒性肿瘤微环境中生存,促进肿瘤生长和免疫逃逸。
在高乳酸微环境中,Tregs上CD36和FoxP3表达水平升高,通过PPAR-β依赖的方式增强FA吸收和FAO,从而增强Tregs的脂毒抗性和代谢适应性(Fig. 3a)。
CD36的消融严重降低了Tregs的脂质摄取,导致肿瘤生长减速。PD-1通过增强ATGL、上调CPT1A和FAO来促进脂肪分解。而抗CD36和抗PD -1联合治疗有协同作用,这表明CD36阻断可能是当前免疫疗法的一种有效的增强剂,并有减少治疗副作用的效果。
考虑到肿瘤浸润Tregs的代谢特征,研究者认为FA合成可能比FA摄取在形成脂质Tregs池和促进其增殖方面更重要。OX40是肿瘤坏死因子受体超家族成员4,它通过促进FA合成,而不是FA摄取来塑造Tregs的脂质构成并促进其增殖。小鼠肿瘤模型和临床数据证实,激活甾醇调节元件结合蛋白可促进脂质合成并支持TME中OX40+ Tregs的局部增殖。
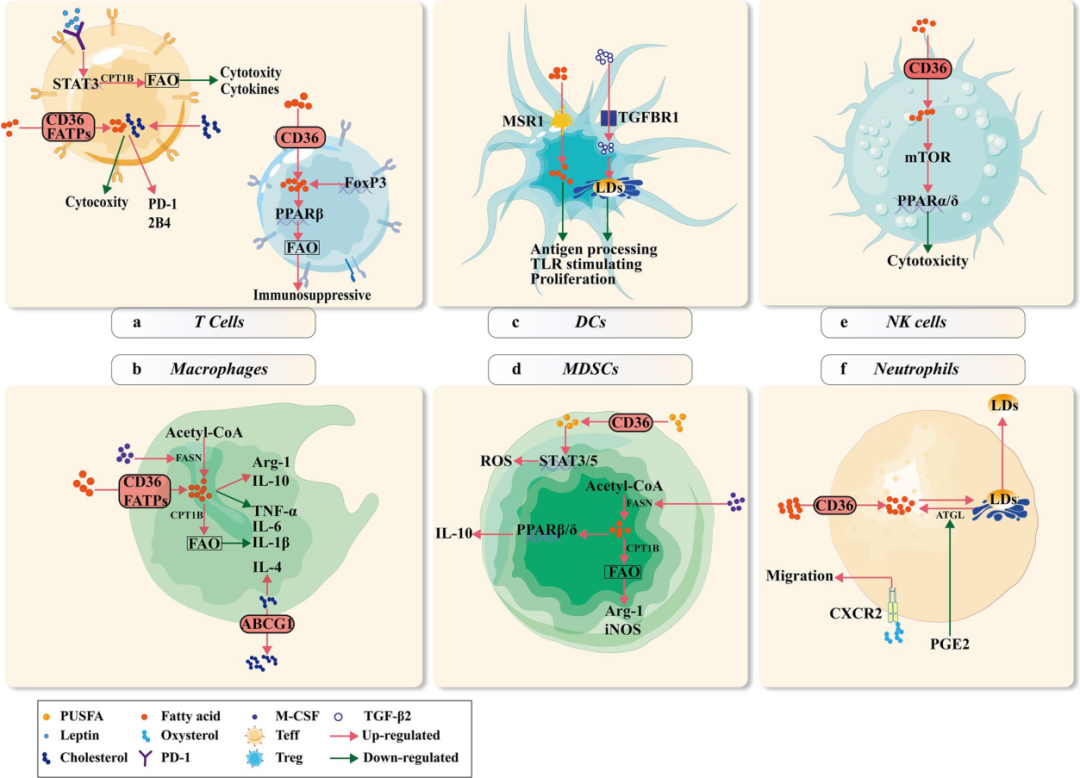
Fig 3 肿瘤前免疫反应中的脂质代谢。通过CD36或FATPs吸收的FAs通过诱导Tefs耗尽或刺激treg中的PPAR-β和FAO介导免疫抑制反应。FoxP3还通过调节treg中的FAs代谢作为一种重要的免疫抑制介质。胆固醇诱导PD-1和2B-4的表达,并随后耗尽Tefs促进肿瘤生长。TME中的Leptin通过PD-1-STAT3-CPT1B通路抑制Tefs,从而增强FAO,消除细胞毒性。b FAs通过转运蛋白摄入或在巨噬细胞内重新合成可刺激CPT1B和FAO,从而增强ARG-1和IL-10等免疫抑制细胞因子的分泌,或抑制TNF-α、IL-6和IL-1β等炎症细胞因子的分泌。来自TME的M-CSF增强FASN表达。高表达ABCG1的巨噬细胞将胆固醇转运到体外,促进IL-4表达和肿瘤进展。c MSR1和TGFBR1促进树突状细胞中FAs转运和LD的形成,从而影响树突状细胞的抗原加工、TLR刺激和增殖。d MDSCs上CD36吸收的PUSFAs激活STAT3/5并刺激ROS的产生。M-CSF促进MDSCs中FASN和FA的产生,进而增加免疫抑制细胞因子的产生,如IL-10、ARG-1和iNOS。e FAs通过mTOR-PPAR信号通路抑制NK细胞的细胞毒性。f由于外源性FAs摄入增加和PGE2下调脂解酶ATGL, ld在肿瘤浸润中性粒细胞中富集。然后将中性粒细胞的ld转移到肿瘤细胞,促进其增殖和进展。羟甾醇通过与CXCR2结合促进中性粒细胞的迁移。
细胞间FAs和胆固醇对TAMs和MDSCs有相反的作用
巨噬细胞和MDSCs都来源于骨髓,并向肿瘤组织迁移。因此,这两种细胞亚型表现出相似的脂质代谢特征。
虽然巨噬细胞参与多种抗肿瘤免疫应答,但肿瘤相关巨噬细胞(tumor-associated macrophages, TAMs)在TME中受多种因素影响,会被重新编程为M2表型,并发挥促肿瘤作用。M1更容易利用糖酵解,而M2主要依赖于FAO。
TAMs倾向于通过CD36从TME中摄取更多的脂质,以维持其促瘤能力。此外,TAMs可以通过乙酰辅酶a的从头合成来满足其对FA的需求。来自Lewis肺癌的巨噬细胞集落刺激因子诱导TAMs中FASN表达,以ARG1和IL-10依赖的方式刺激肿瘤生长和血管生成(Fig. 3b)。FASN的抑制使TAMs中TNF-α、IL-6、IL-10和ROS的表达被显著抑制,从而削弱其促肿瘤活性。这些结果表明,异常的脂质积累是TAMs参与肿瘤前免疫应答的必要条件。
α / β -水解酶结构域蛋白(TAG的脂解因子)和MAGL(又称MGLL,单酰基甘油的关键脂肪酶)是调节脂质代谢平衡的关键酶。MGLL过表达小鼠模型中TAMs的脂质积累完全受到抑制,同时促炎细胞因子(IL-1β、TNF-α和IFN-γ)表达水平升高,抗炎细胞因子(IL-10、ARG-1、转化生长因子-β (TGF-β)和IL-4)表达水平降低。TGF-β通过激活Mek1/2、Erk1/2和Rsk1/2促进LD的形成,进一步增强TAMs的免疫抑制能力。
Mek的抑制作用可以抑制LD的形成,促使细胞因子的平衡向M1表型转移,避免ROS和NO水平过多的降低。需要指出的是,在TAMs中,FA的分解代谢也会被加强,以产生更多的能量,维持前肿瘤表型。升高的CTP1A通过调节JAK1-STAT6轴与升高的FAO、ATP生产和氧化应激启动和形成TAMs。用依托莫西抑制FAO,可以通过降低TAMs的吞噬能力和下调IL-1β的表达,以ROS-NLRP3依赖的方式逆转TAMs的促肿瘤作用(Fig. 3b)。
此外,敲除受体相互作用蛋白激酶3,可通过增强FAO和氧化磷酸化相关基因(Cpt1a、Cpt1b、Acadvl和Hadh)来促进M2表型,尤其是增强巨噬细胞中PD-L1的表达。这提示我们FAs在TAMs中具有抗炎作用。
TME中的MDSCs表现出强大而全面的免疫抑制作用,它不仅能通过促进肿瘤细胞生长来表现免疫抑制作用,还能通过与其他免疫细胞建立复杂的网络来表现。
通过转运受体(Slc27a1/Fatp1, Slc27a6/Fatp6, Msr1, CD36和ldlr)的脂质摄取增加而导致的不可控脂质积累是肿瘤浸润性MDSCs代谢重编程的原因。不饱和脂肪酸(USFAs)促进细胞内LD的形成,抑制MDSCs而非SFAs的活性。
(α-)亚麻酸(USFA)通过激活JAK-STAT3和ROS,显著促进多形核MDSCs的扩张和比例,并对T细胞产生比SFAs作用更强的抑制作用(Fig. 3d)。CD36基因缺失或STAT3/5基因抑制抑制了MDSCs的氧化代谢和免疫抑制功能,导致ARG1和iNOS减少,进而导致CD8+ T细胞依赖的肿瘤免疫反应延迟。
与TAMs相似,来自Lewis肺癌细胞的巨噬细胞集束刺激因子也可以提高MDSCs中FASN的表达,从而激活PPAR-β/δ以及随后IL-10的分泌(Fig. 3d)。此外,心磷脂可以通过激活MDSCs中的PPAR-γ来刺激IL-10的分泌,而MDSCs驻留在荷瘤小鼠的肺中。
MDSCs更多地依赖于FAO来维持其免疫抑制和肿瘤促进能力。在单核细胞MDSCs和多形核MDSCs中均观察到CPT1和3-羟基酰基- CoA脱氢酶的高表达,免疫抑制因子(ARG1、iNOS2和NO)的表达增加,以及T细胞增殖率和IFN的产生降低(Fig. 3d)。
FAO抑制剂依托莫西可通过抑制MDSCs的浸润,增强过继T细胞治疗和低剂量化疗的抗肿瘤作用。这表明靶向FAO可以限制MDSCs的免疫抑制功能,促进其他抗肿瘤治疗。
与FAs不同,胆固醇在TAMs和MDSCs中是一种炎症介质。ABCG1在TAMs中高度表达,促进胆固醇外排率和随后的IL-4分泌,以维持其抗炎特性(Fig. 3b)。在来自西方样饮食喂养的Abcg1−/−小鼠肿瘤组织中的TAMs显示出较高的凋亡率,更多细胞从M2表型向M1表型转变,对肿瘤细胞的细胞毒性增强,肿瘤生长降低。
此外,巨噬细胞缺乏ABCG1可改变固有细胞因子的产生,增加NK细胞和CD4+T细胞在TME中的浸润,阻止肿瘤生长。然而,细胞色素P450家族27亚家族A成员1 (CYP27A1)是将胆固醇转化为27-羟基胆固醇(27HC)所需的一种细胞色素P450氧化酶,在乳腺肿瘤浸润的巨噬细胞上高表达。
这意味着来自巨噬细胞的27HC是乳腺肿瘤生长的促进因子。当CYP27A1被抑制时,这种促肿瘤作用被削弱。其机制可能是27HC或其他胆固醇前体和羟甾醇通过激活LXRs使巨噬细胞极化为M2表型。
到目前为止,关于MDSCs胆固醇代谢的研究还很有限。有研究认为,在髓系细胞中缺失ABCA1/ G1基因可阻断其胆固醇流出,限制MDSCs和肿瘤生长。
LXR激动剂通过LXR β核激素受体和载脂蛋白E (ApoE)刺激胆固醇外排,可以显著降低体外和体内MDSCs的比例。在ApoE缺失的荷瘤小鼠中,循环和肿瘤内的MDSCs高于对照组,会导致肿瘤生长更快。
此外,髓系细胞中PD-1缺失导致Erk1/2和哺乳动物雷帕霉素靶蛋白(mTOR) C1在粒细胞集落刺激因子作用下被激活,促进胆固醇合成和髓系细胞的分化。这导致了有效的先天和适应性抗肿瘤反应。
这些发现提示MDSCs和TAMs中胆固醇含量的增加可能协同激活抗肿瘤免疫反应,消除恶性肿瘤。脂质在巨噬细胞中的不同作用可能与FAs、胆固醇和恶性肿瘤的亚型有关。
因此,针对巨噬细胞脂质代谢的进一步临床应用应考虑这些矛盾的结果,开发更具体的脂质靶向治疗,以最大限度地发挥其抗肿瘤作用。
高脂含量损害树突状细胞的抗原呈递能力
树突状细胞的抗肿瘤作用主要来源于其抗原提呈能力。虽然高脂含量使肝脏DCs具有更强的交叉提呈能力,但许多研究表明FAs也可能是肿瘤浸润性DCs中的免疫抑制因子。
饮食诱导的肥胖导致共刺激分子和DC相关细胞因子表达受限,从而诱导DCs在荷瘤小鼠中激活T细胞。此外,在辐射诱导的胸腺淋巴瘤中,脂蛋白脂肪酶和FABP1水平升高导致血清TAG水平升高,并导致DC功能障碍。这些结果表明,脂质倾向于介导来自DCs的前肿瘤免疫反应。
此外,TME内的可溶性因子通过上调树突状细胞氧化中性脂质含量,限制了MHC I类复合物的形成,削弱了树突状细胞的抗原呈递能力。同时,SFAs和PUSFAs显著降低DCs的分化和激活,进而降低T细胞功能。
巨噬细胞清偿剂受体1 (Macrophage scavenger receptor 1, MSR1)是导致DC细胞中FA水平升高和CD8+ T细胞被诱导能力受限的原因(Fig. 3c)。阻断MSR1会削弱DCs中FA的积累,从而刺激过从性反式转化的肿瘤特异性CD8+ T细胞在乳腺肿瘤中的扩张和细胞毒性。
另一种脂质摄取蛋白类固醇受体RNA激活因子1也影响DCs的抗原呈递能力和免疫原性。阻断树突状细胞FA摄取或损害树突状细胞脂质合成可通过提高T细胞的受刺激能力来减弱癌细胞的免疫抑制状态。
此外,Zhao等发现黑素瘤可以通过Wnt5a-β-catenin PPAR-γ信号通路上调CPT1A的表达,增强DCs中的FAO。这种转移增强了吲哚胺2,3-双氧合酶-1的数量和活性,同时抑制了IL-6和IL-12的表达,最终导致Treg的产生。
树突状细胞脂质代谢障碍会导致LD积累。LD通过降低MHC表达和Ag特异性T细胞的刺激能力,来削弱DCs的抗原呈递能力。
在酸性TME中,自分泌TGF-β2导致LD在DCs中异常积累,抑制其向淋巴结的增殖和迁移能力以及CD8+ T细胞刺激能力(Fig. 3c)。而SB‐431542是一种有效的TGF-β I受体抑制剂,可减弱TGF-β引起的DC疫苗的抑制作用。
本研究提示,小分子抑制剂可能通过影响脂质代谢来调节肿瘤免疫。基于以上讨论,可能是由于这些异常积累的脂质所处的器官或脂质种类的不同,树突状细胞内脂质积累的确切作用尚不清楚。
根据树突状细胞的标记和形态,将树突状细胞分为髓系树突状细胞和浆细胞样树突状细胞。这两种亚型是否具有不同的脂代谢模式目前尚不清楚。进一步的研究应侧重于检测肿瘤相关树突状细胞脂质代谢的具体机制。
脂质在其他免疫细胞中的免疫抑制作用
NK细胞通过产生穿孔素、GzmB等细胞毒性颗粒或IFN-γ、TNF等炎症细胞因子,直接杀伤肿瘤细胞,是抗肿瘤免疫反应中快速有效的应答者。TME中的许多因子都会导致NK细胞从糖酵解到氧化磷酸化的转换,使其抗肿瘤特性失活,导致肿瘤免疫逃逸。
富含脂质的TME迫使肿瘤浸润的NK细胞摄取更多的脂质,从而抑制细胞毒性因子的流动,及由于“代谢麻痹”在体内和体外产生的抗肿瘤功能。转录分析表明,高水平的mTOR可以刺激PPAR -α/δ目标基因,包括影响参与脂质和甘油吸收(Cd36, Lpl和Lrp4)的基因,影响LD形成和使脂酶(时间和Plin2)及脂质代谢(Abca1、Scarb2 Gyk)在高脂肪饮食小鼠NK细胞中,表现出低细胞毒性(Fig. 3e)。
mTOR是细胞代谢的主要调节因子,调控NK细胞的发育和活化。敲除NK细胞中CISH基因可刺激mTOR信号通路,增加NK细胞代谢适应度,进而提高其抗肿瘤能力。这些结果表明,肥胖可以通过mTOR-PPAR通路来影响肿瘤相关NK细胞。因此,PPAR靶向治疗可以通过减少肿瘤细胞增殖和激活NK细胞能力的同时,消除肿瘤生长。
除实体瘤外,来自弥漫性大B细胞淋巴瘤个体的NK细胞中脂质代谢增加,IFN-γ表达降低。转录分析显示,在弥漫性大B细胞淋巴瘤小鼠中,脂质代谢相关基因,包括Cd36、Fabp4、Fabp5和Pparg出现富集,这与中性脂质水平的显著升高相一致。体内实验也证实,在较低浓度的FAs组合的模拟生理条件下,IFN-γ和GzmB显著降低。
一个有趣的现象是,荷瘤小鼠的手术治疗可上调CD36高表达、GzmB低表达的脾NK细胞脂质含量。这一结果提示,恶性肿瘤手术后的脂质供应可能对抗肿瘤免疫反应产生不良影响。
综上所述,我们可以从上述讨论中推断,胆固醇对NK细胞的抗肿瘤能力有促进作用,而FA的积累则可能没有促进作用。这意味着在NK细胞靶向治疗中,抑制FAs和促进胆固醇的共同作用可能会导致抗肿瘤免疫反应的增强。
Ns也被发现可能参与肿瘤相关的免疫反应。与吞噬细菌、诱导组织损伤、在感染中激活免疫系统等正常能力相反,肿瘤相关的Ns被认为参与了肿瘤前免疫应答。
人们普遍认为,Ns通过加速弥散性肿瘤细胞的迁移和增殖,并通过中性粒细胞胞外陷阱唤醒它们,来促进肿瘤细胞向肺转移。4T1肿瘤模型肺转移前组织中,Ns高表达与脂质吸收和LD形成相关基因,低表达与参与LD降解和β-氧化的相关基因。这意味着,肺内Ns具有脂质积聚的潜力,这种潜力会癌症进展过程中增强。
同时,来自间充质细胞的前列腺素E2通过激活Hilpda抑制ATGL,促进Ns中LD的形成。然后这些LDs被转移到癌细胞中,促进肿瘤转移(Fig. 3f)。与野生型小鼠相比,Atgl−/−小鼠表现出更强的自发肺转移,但原发肿瘤生长没有改变。同时,胆固醇也会影响肿瘤浸润的Ns。
肿瘤来源的羟甾醇通过CXCR2依赖途径招募肿瘤前Ns,促进新血管生成和免疫抑制。百日流毒素抑制CD11bhigh Gr1high Ly6G+细胞向22(R)-羟基胆固醇的迁移,这表明G蛋白偶联受体介导这些细胞向LXR配体的迁移,并随后激活CXCR2信号(Fig. 3f)。
当在荷瘤小鼠中,阻断羟甾醇CXCR2轴和损害中性粒细胞迁移时,会出现更好的延迟肿瘤生长和延长总生存的效果。进一步研究肿瘤浸润Ns中的脂质代谢,有望为恶性肿瘤的治疗,特别是在限制转移方面,开辟新的途径。
脂质代谢与肿瘤免疫治疗
近年来的研究揭示了脂质代谢在肿瘤发生、进展和转移以及调节肿瘤免疫中的重要作用。有研究者提出了利用靶向肿瘤微环境中异常脂质代谢的癌症治疗策略。而鉴于各种已被开发利用于治疗心血管疾病的针对脂质代谢的抑制剂,重新利用现有药物,针对癌症中的异常脂质代谢的方法可能是有效的。然而,需要注意的是,针对脂质代谢的治疗可能同时影响肿瘤细胞和免疫细胞。迄今为止,许多针对肿瘤脂质代谢的治疗在增强抗肿瘤免疫应答方面取得了显著成效。
鉴于外源性FA的摄取在很大程度上依赖于CD36的表达,在临床前研究中应用CD36抑制剂可阻碍多种癌症的生长,并在与FASN抑制剂和抗PD -1疗法结合时显示协同作用。
抑制CD36除了可以直接消除肿瘤细胞外,还可以通过消除肿瘤内Tregs的功能来细化免疫抑制TME,抑制肿瘤进展。同时,用单克隆抗体阻断DCs上的CD36,使其MHC II类分子的抗原呈递能力减弱,从而改善CD4+ T细胞启动,进而增强其抗肿瘤免疫反应(Table 2)。此外,沉默DCs上的MSR1可提高DC的接种效果。
除脂肪酸吸收外,脂肪酸从头合成也占脂肪来源的很大比例。ACLY将柠檬酸盐和辅酶a转化为乙酰辅酶a和草酰乙酸盐,将糖酵解和脂类合成连接在一起。ACC催化乙酰辅酶a向丙二酰辅酶a的转化。通过抑制剂或siRNA干扰其表达,进而诱导肿瘤凋亡,限制肿瘤生长,并协同增强化疗或靶向药物。
腺苷5 ‘单磷酸活化蛋白激酶(Adenosine 5 ‘ monophospe -activated protein kinase, AMPK)是ACC的关键抑制因子,二甲双胍是AMPK的典型激动剂。AMPK除直接影响肿瘤细胞外,还可能影响肿瘤相关免疫细胞的脂质代谢。一项II期临床试验报道,低剂量二甲双胍的应用增加了TME中CD8+T细胞的数量并减少了肿瘤相关的巨噬细胞(Table 2)。
然而,最近的一项研究报道,在巨噬细胞中消耗ACLY并不能增强小鼠的抗肿瘤免疫应答。硬脂酰辅酶a去饱和酶(SCD)是FA合成的另一种关键酶,与肿瘤发生、肿瘤进展和总生存期相关。SCD1抑制剂和靶向治疗在癌症中发挥协同抑制作用。虽然SCD在炎症性疾病中发挥作用,但尚无直接证据显示其与肿瘤免疫的关系。
来自FASN催化的Acetoacetyl-CoA将FA的生物合成与胆固醇的生物合成联系起来,促进脂筏和Toll样受体(TLR) 4信号通路的形成,激活巨噬细胞中更高水平的细胞因子反应。FASN抑制剂C75可以抑制小鼠骨髓源性巨噬细胞脂质合成,可降低IL-1β、IL-10、IL-6、和TNF-α水平对各种TLR激动剂的响应(Table 2)。在临床前研究中,一些针对FASN的新化合物已经开发出来,它们具有较轻的副作用和有效的抗肿瘤作用。因此,进一步分析这些药物对肿瘤免疫的影响是必要的。
FA分解在肿瘤相关免疫细胞中也很重要。具体来说,抑制巨噬细胞中二酰基甘油酰基转移酶(A922500, mf -06424439)、ATGL (SML1075)和MAGL (SML0872)的催化活性会减弱其免疫抑制能力。靶向CPT1的疗法发挥抗肿瘤作用,例如减少肝细胞癌细胞中invadopodia的形成并消除TAM在体内和体内的促肿瘤作用(Table 2)。
etomoxir抑制FAO对TME中F4/80+巨噬细胞的数量没有影响,但降低了F4/80+巨噬细胞对胰管腺癌细胞的吞噬。在其他类型的免疫抑制细胞中,依托莫西与过从T细胞治疗和低剂量化疗具有协同作用,可以抑制肿瘤浸润的MDSCs,提高抗肿瘤疗效。另一种FAO抑制剂perhexiline可使乳腺癌干细胞对化疗重新敏感,并增强CD8+ Tefs的功能。
所有这些研究都为肿瘤相关免疫细胞中FA代谢的干扰提供了有力的证据,同时证明可以通过该途径回避肿瘤细胞对免疫系统的免疫抑制作用。肿瘤相关的免疫细胞一旦从代谢阻滞中释放出来,就会从前肿瘤表型转变为抗肿瘤表型,并单独或与其他疗法联合表现出增强的肿瘤清除能力。
除了FAs,胆固醇也是肿瘤免疫治疗研究中的重要脂质。LXRs是膳食胆固醇的主要传感器,通过增加胆固醇的排泄和减少胆固醇的吸收,转录调节胆固醇稳态。LXR激动剂可有效诱导小鼠模型肿瘤消退并延长小鼠生存。在免疫小鼠模型中,LXR激活显示出明显的抗肿瘤作用,这表明免疫细胞参与了LXR激动诱导的抗肿瘤活性。LXR激动剂GW3965与一线药物达卡巴嗪和抗CTLA-4抗体联合使用时表现出加性抗肿瘤效果。RGX-104激活LXRs,通过上调促凋亡Bcl-2家族成员的表达抑制MDSCs的生存和免疫抑制功能(Table 2)。
由于细胞膜中的游离胆固醇可以促进T细胞的免疫应答,使用avasimibe抑制ACAT在使用或不使用抗PD -1治疗的情况下,在减少肿瘤进展中有深远的疗效(Table 2)。avasimibe和chemo联合应用也能增强B16F10黑素瘤异种移植及肺转移模型中CTL和PTX/αGC-TH-Lip的抗肿瘤能力(Table 2)。
由于其物理化学和生物特性,脂质除了能作为能量来源或物理调节器外,还可以被改造为有效的转运体。迄今为止,脂基囊泡,包括脂质体、固体脂基系统、脂质颗粒、非离子表面活性剂囊泡和胶束,均已被开发为药物载体,能提高药物传递效率,并在临床前和临床研究中取得了令人满意的结果。
脂质体是第一种用于癌症治疗的脂质纳米颗粒。脂质体具有较高的生物相容性和生物降解性,可提供多种靶向特异性高的有效载荷,有助于化疗药物进入TME,并限制药物严重的副作用。Doxil®是FDA批准的第一种用脂质体包封阿霉素的纳米药物,它可以利用其特异性进行肿瘤定位,并在肿瘤部位释放阿霉素。
此外,使用脂质体或其他脂质基纳米颗粒作为小干扰RNA的载体,可以克服因其在血液中的不稳定性和负电荷密度而导致的临床用药障碍,这是一种很有希望的运用于治疗顽固性疾病的武器。现已进行了几项临床试验来观察其疗效和副作用。
因此,尽管FA和胆固醇代谢在肿瘤相关的免疫细胞中很重要,但相应的治疗方法仍然缺乏。由于脂质代谢具有明显的影响在不同的肿瘤相关的免疫细胞,甚至有负面影响在同一细胞类型由于恶性肿瘤的类型和脂质,这些复杂的现象和机制需要新颖的治疗策略,目标在肿瘤相关的免疫细胞脂质代谢。更具体地说,针对不同恶性肿瘤中特定的脂质亚型、酶和免疫细胞可能是开发脂质相关抗肿瘤药物的一个有前景的方向。
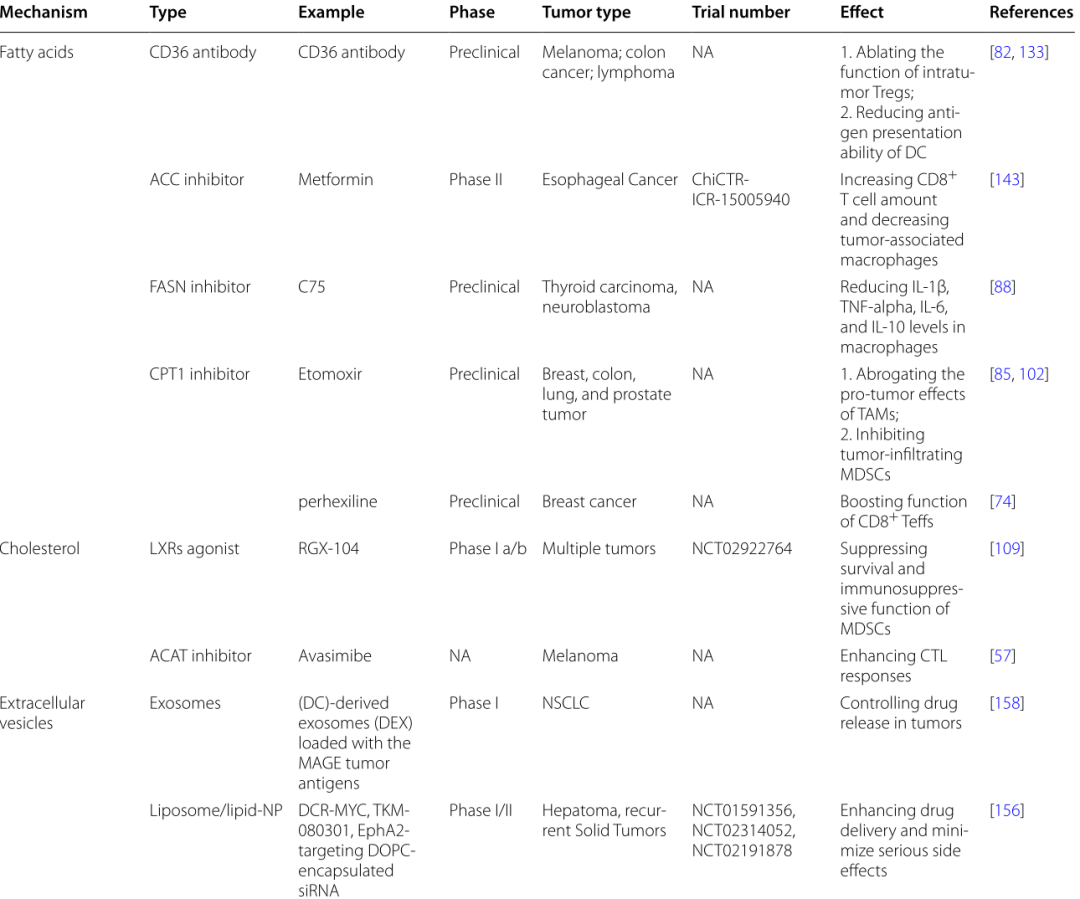
Table 2 肿瘤浸润性免疫细胞脂质代谢的策略
总结与展望
脂质代谢重编程现在被认为是恶性肿瘤的一个重要特征。在缺氧、酸性和营养缺乏的TME中,癌症和免疫细胞往往依赖脂质来储存能量、形成细胞膜的细胞构建块和信号分子来源。因此,TME中异常调节的脂质可显著影响肿瘤的发生、后续发展和转移。
同时,TME内的肿瘤相关免疫细胞也受到这些普遍存在的脂质生物分子的影响。TME中的肿瘤浸润免疫细胞根据其在肿瘤相关免疫应答中的作用可分为抗肿瘤表型和促肿瘤表型。在TME中,脂质是一把双刃剑,既支持抗肿瘤免疫反应,也支持促肿瘤免疫反应。
例如,促进FAO可以中和CD8+ T细胞中PD-1抗体的抑制作用,增强CD36表达有助于维持CD8+ T细胞的有效功能和长期生存。CAR-T细胞中FA代谢的改变可以改善其抗肿瘤免疫能力。但是,也有报道称,过多的FAs会抑制TME中Tefs的抗肿瘤能力,支持Tregs的增殖和免疫抑制功能。
这些矛盾的结果也可以在其他免疫细胞中发现,而这些矛盾结果使我们处以一个两难的困境,因为无法通过单纯抑制或刺激TME中的一种脂质代谢途径获得最佳结果。目前针对肿瘤浸润免疫细胞脂质代谢的治疗多集中在单一类型的细胞或代谢途径上,但这是远远不够的。这意味着为了在恶性肿瘤患者中获得更合理、更有效的结果,在未来针对脂质代谢的靶向治疗中应该考虑到这些矛盾。
综上所述,我们综述了大量关于TME内脂质代谢及其相关信号通路的研究,这些研究对多种类型的恶性肿瘤产生了多样而深刻的影响。然而,有关的具体机制仍有待阐明。需要进一步的基础研究和药物开发来阐明脂质在肿瘤免疫学中的作用,并优化现有的癌症治疗方法。
— THE END —
▉ 强烈推荐
本公众号上的往期文章同步发布至对应网站OncoLab实验室。
网站自带检索功能,可以根据关键词进行检索,并且可以根据日期及内容分类进行查看 ,大家可以收藏方便在电脑上查看。
网址是:oncolab.cn
此外,梳理了一下这几年攒的收藏夹,做了一个导航网页,包含常用网站、文献阅读、试剂订购、基金相关、实用工具、常用数据库等分类内容,并且整合了百度、谷歌、必应三大搜索引擎到检索工具中,欢迎收藏或设置为主页使用~
网址是:dh.oncolab.cn
关注本号~

加入读者交流群~

加入知识星球~
用于OncoLab读者交流互助永久免费
本篇文章来源于微信公众号: OncoLab
 微信扫一扫打赏
微信扫一扫打赏
 支付宝扫一扫打赏
支付宝扫一扫打赏